Xeloda 500mg Capecitabine Verwendung, Nebenwirkungen, Stärke und Dosierung. Preis in Online-Apotheke. Generika medikamente rezeptfrei.
Was ist Xeloda 500 mg und wie wird es angewendet?
Xeloda ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Krebserkrankungen wie Dickdarmkrebs, Dickdarmkrebs und Brustkrebs. Xeloda kann allein oder zusammen mit anderen Medikamenten angewendet werden.
Xeloda gehört zu einer Arzneimittelklasse namens Antineoplastika, Antimetaboliten.
Es ist nicht bekannt, ob Xeloda 500 mg bei Kindern sicher und wirksam ist.
Welche Nebenwirkungen kann Xeloda 500 mg haben?
Xeloda kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Xeloda 500 mg gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Xeloda. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
WARNUNG
XELODA-WARFARIN-INTERAKTION
Wechselwirkungen mit XELODA 500 mg Warfarin: Bei Patienten, die gleichzeitig mit Capecitabin und einem oralen Cumarinderivat als Antikoagulans behandelt werden, sollte das Ansprechen auf das Antikoagulans (INR oder Prothrombinzeit) häufig überwacht werden, um die Dosis des Antikoagulans entsprechend anzupassen. Eine klinisch bedeutsame Arzneimittelwechselwirkung zwischen XELODA und Warfarin wurde in einer klinisch-pharmakologischen Studie nachgewiesen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und ARZNEIMITTELWECHSELWIRKUNGEN]. Veränderte Gerinnungsparameter und/oder Blutungen, einschließlich Tod, wurden bei Patienten berichtet, die XELODA 500 mg gleichzeitig mit Antikoagulanzien auf Cumarinbasis wie Warfarin und Phenprocoumon einnahmen. Postmarketing-Berichte haben klinisch signifikante Verlängerungen der Prothrombinzeit (PT) und der INR bei Patienten gezeigt, die zum Zeitpunkt der Einführung von XELODA 500 mg mit Antikoagulanzien stabilisiert waren. Diese Ereignisse traten innerhalb von mehreren Tagen und bis zu mehreren Monaten nach Beginn der Behandlung mit XELODA 500 mg und in einigen Fällen innerhalb von 1 Monat nach Beendigung der Behandlung mit XELODA auf. Diese Ereignisse traten bei Patienten mit und ohne Lebermetastasen auf. Ein Alter über 60 und eine Krebsdiagnose prädisponieren unabhängig voneinander für ein erhöhtes Risiko einer Koagulopathie.
BEZEICHNUNG
XELODA (Capecitabin) ist ein Fluorpyrimidincarbamat mit antineoplastischer Aktivität. Es ist ein oral verabreichtes systemisches Prodrug von 5'-Desoxy-5-Fluoruridin (5'-DFUR), das in 5-Fluorouracil umgewandelt wird.
Der chemische Name für Capecitabin lautet 5'-Desoxy-5-Fluor-N-[(Pentyloxy)-Carbonyl]-Cytidin und hat ein Molekulargewicht von 359,35. Capecitabin hat die folgende Strukturformel:
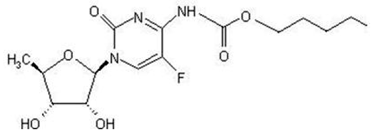
Capecitabin ist ein weißes bis cremefarbenes kristallines Pulver mit einer Wasserlöslichkeit von 26 mg/ml bei 20 °C.
XELODA wird als bikonvexe, längliche Filmtabletten zur oralen Verabreichung geliefert. Jede hell pfirsichfarbene Tablette enthält 150 mg Capecitabin und jede pfirsichfarbene Tablette enthält 500 mg Capecitabin. Zu den inaktiven Bestandteilen in XELODA gehören: wasserfreie Lactose, Croscarmellose-Natrium, Hydroxypropylmethylcellulose, mikrokristalline Cellulose, Magnesiumstearat und gereinigtes Wasser. Die pfirsichfarbene oder hellpfirsichfarbene Filmbeschichtung enthält Hydroxypropylmethylcellulose, Talk, Titandioxid und synthetische gelbe und rote Eisenoxide.
INDIKATIONEN
Darmkrebs
Brustkrebs
DOSIERUNG UND ANWENDUNG
Wichtige Verwaltungsanweisungen
XELODA Tabletten sollten unzerkaut mit Wasser innerhalb von 30 Minuten nach einer Mahlzeit geschluckt werden. XELODA 500 mg ist ein Zytostatikum. Befolgen Sie die geltenden besonderen Handhabungs- und Entsorgungsverfahren.1 Wenn XELODA 500 mg Tabletten geschnitten oder zerkleinert werden müssen, sollte dies von einem Fachmann durchgeführt werden, der im sicheren Umgang mit zytotoxischen Arzneimitteln unter Verwendung geeigneter Ausrüstung und Sicherheitsverfahren geschult ist. Die XELODA-Dosis wird anhand der Körperoberfläche berechnet.
Standard-Anfangsdosis
Monotherapie (metastasierter Darmkrebs, adjuvanter Darmkrebs, metastasierter Brustkrebs)
Die empfohlene Dosis von XELODA beträgt 1250 mg/m2 oral zweimal täglich (morgens und abends; entspricht einer Tagesgesamtdosis von 2500 mg/m2) über 2 Wochen, gefolgt von einer einwöchigen Ruhephase, die als 3-wöchige Zyklen gegeben wird (siehe ).
Eine adjuvante Behandlung bei Patienten mit Dickdarmkrebs von Dukes C wird für insgesamt 6 Monate empfohlen [dh XELODA 1250 mg/m2 oral zweimal täglich für 2 Wochen, gefolgt von einer einwöchigen Ruhephase, gegeben als insgesamt 3-wöchige Zyklen von 8 Zyklen (24 Wochen)].
In Kombination mit Docetaxel (metastasierter Brustkrebs)
In Kombination mit Docetaxel beträgt die empfohlene Dosis von XELODA 1250 mg/m2 zweimal täglich für 2 Wochen, gefolgt von einer 1-wöchigen Ruhephase, kombiniert mit 75 mg/m2 Docetaxel als 1-stündige intravenöse Infusion alle 3 Wochen. Bei Patienten, die die Kombination XELODA plus Docetaxel erhalten, sollte gemäß der Docetaxel-Kennzeichnung vor der Verabreichung von Docetaxel mit einer Prämedikation begonnen werden. zeigt die tägliche Gesamtdosis von XELODA nach Körperoberfläche und die Anzahl der Tabletten, die bei jeder Dosis eingenommen werden müssen.
Richtlinien zum Dosismanagement
Allgemein
Die XELODA-Dosierung muss möglicherweise individuell angepasst werden, um das Patientenmanagement zu optimieren. Die Patienten sollten sorgfältig auf Toxizität überwacht werden, und die Dosen von XELODA 500 mg sollten nach Bedarf angepasst werden, um der individuellen Verträglichkeit der Behandlung durch den Patienten Rechnung zu tragen [siehe Klinische Studien ]. Toxizität aufgrund der Verabreichung von XELODA 500 mg kann durch symptomatische Behandlung, Dosisunterbrechungen und Anpassung der XELODA-Dosis behandelt werden. Nachdem die Dosis reduziert wurde, sollte sie zu einem späteren Zeitpunkt nicht mehr erhöht werden. Dosen von XELODA 500 mg, die wegen Toxizität ausgelassen wurden, werden nicht ersetzt oder wiederhergestellt; Stattdessen sollte der Patient die geplanten Behandlungszyklen wieder aufnehmen.
Die Dosis von Phenytoin und die Dosis von Cumarinderivat-Antikoagulanzien müssen möglicherweise reduziert werden, wenn eines der beiden Arzneimittel gleichzeitig mit XELODA verabreicht wird [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Monotherapie (metastasierter Darmkrebs, adjuvanter Darmkrebs, metastasierter Brustkrebs)
Für das Management von Nebenwirkungen wird das unten beschriebene XELODA-Dosismodifikationsschema (siehe ) empfohlen.
In Kombination mit Docetaxel (metastasierter Brustkrebs)
Dosismodifikationen von XELODA 500 mg wegen Toxizität sollten wie oben für XELODA beschrieben vorgenommen werden. Wenn zu Beginn eines Behandlungszyklus eine Behandlungsverzögerung entweder für XELODA 500 mg oder für Docetaxel indiziert ist, sollte die Verabreichung beider Wirkstoffe verschoben werden, bis die Voraussetzungen für die Wiederaufnahme beider Arzneimittel erfüllt sind.
Das Dosisreduktionsschema für Docetaxel bei Anwendung in Kombination mit XELODA zur Behandlung von metastasiertem Brustkrebs ist in Tabelle 3 dargestellt.
Anpassung der Anfangsdosis bei speziellen Patientengruppen
Nierenfunktionsstörung
Bei Patienten mit leichter Nierenfunktionsstörung (Kreatinin-Clearance = 51 bis 80 ml/min [Cockroft und Gault, wie unten gezeigt]) wird keine Anpassung der Anfangsdosis von XELODA 500 mg empfohlen. Bei Patienten mit mäßig eingeschränkter Nierenfunktion (Kreatinin-Clearance zu Studienbeginn = 30 bis 50 ml/min) sollte eine Dosisreduktion auf 75 % der XELODA-Anfangsdosis bei Anwendung als Monotherapie oder in Kombination mit Docetaxel (von 1250 mg/m2 auf 950 mg/m2) empfohlen werden zweimal täglich) wird empfohlen [vgl Verwendung in bestimmten Bevölkerungsgruppen und KLINISCHE PHARMAKOLOGIE ]. Eine anschließende Dosisanpassung wird empfohlen, wie in beschrieben und (je nach Behandlungsschema), wenn ein Patient ein unerwünschtes Ereignis vom Grad 2 bis 4 entwickelt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Empfehlungen zur Anpassung der Anfangsdosis für Patienten mit mäßig eingeschränkter Nierenfunktion gelten sowohl für XELODA 500 mg als Monotherapie als auch für XELODA 500 mg in Kombination mit Docetaxel.
Cockroft- und Gault-Gleichung:
Geriatrie
Ärzte sollten bei der Überwachung der Wirkungen von XELODA 500 mg bei älteren Patienten Vorsicht walten lassen. Für eine Dosierungsempfehlung liegen keine ausreichenden Daten vor.
WIE GELIEFERT
Stärken der Dosierungsformen
XELODA wird als bikonvexe, längliche Filmtabletten zur oralen Verabreichung geliefert. Jede hell pfirsichfarbene Tablette enthält 150 mg Capecitabin und jede pfirsichfarbene Tablette enthält 500 mg Capecitabin.
150mg
Farbe: Heller Pfirsich Gravur: XELODA auf der einen Seite und 150 auf der anderen 150-mg-Tabletten sind in Flaschen zu 60 ( NDC 0004-1100-20), einzeln im Karton verpackt.
500mg
Farbe: Pfirsich Gravur: XELODA 500 mg auf der einen Seite und 500 auf der anderen 500-mg-Tabletten sind in Flaschen zu 120 ( NDC 0004-1101-50), einzeln im Karton verpackt.
Lagerung und Handhabung
Bei 25 °C (77 °F) lagern; Ausflüge erlaubt bis 15° bis 30°C (59° bis 86°F). [Siehe USP Kontrollierte Raumtemperatur]. FEST GESCHLOSSEN HALTEN.
XELODA ist ein Zytostatikum. Befolgen Sie die anwendbaren speziellen Handhabungs- und Entsorgungsverfahren.1 Nicht verwendetes Produkt sollte gemäß den örtlichen Vorschriften oder Medikamentenrücknahmeprogrammen entsorgt werden.
VERWEISE
1. „Gefährliche OSHA-Drogen“. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Vertrieb durch: Genentech USA, Inc. Ein Mitglied der Roche-Gruppe, 1 DNA Way, South San Francisco, CA 94080-4990. Überarbeitet: Mai 2021
NEBENWIRKUNGEN
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Adjuvans Darmkrebs
zeigt die Nebenwirkungen, die bei ≥5 % der Patienten aus einer Phase-3-Studie bei Patienten mit Dukes-C-Darmkrebs auftraten, die mindestens eine Dosis der Studienmedikation erhielten und mindestens einer Sicherheitsbewertung unterzogen wurden. Insgesamt 995 Patienten wurden mit 1250 mg/m2 zweimal täglich XELODA 500 mg behandelt, verabreicht über 2 Wochen, gefolgt von einer 1-wöchigen Ruhephase, und 974 Patienten erhielten 5-FU und Leucovorin (20 mg/m2 Leucovorin i.v., gefolgt von 425 mg/m2 IV Bolus 5FU an den Tagen 1-5 alle 28 Tage). Die mediane Behandlungsdauer betrug 164 Tage bei den mit Capecitabin behandelten Patienten und 145 Tage bei den mit 5-FU/LV behandelten Patienten. Insgesamt 112 (11 %) bzw. 73 (7 %) der mit Capecitabin bzw. 5-FU/LV behandelten Patienten brachen die Behandlung aufgrund von Nebenwirkungen ab. Insgesamt 18 Todesfälle aus allen möglichen Gründen traten entweder während der Studie oder innerhalb von 28 Tagen nach Erhalt des Studienmedikaments auf: 8 (0,8 %) Patienten wurden randomisiert zu XELODA 500 mg und 10 (1,0 %) randomisiert zu 5-FU/LV.
zeigt Laboranomalien Grad 3/4, die bei ≥ 1 % der Patienten aus einer Phase-3-Studie bei Patienten mit Dickdarmkrebs von Dukes C auftraten, die mindestens eine Dosis der Studienmedikation erhielten und mindestens einer Sicherheitsbewertung unterzogen wurden.
Metastasierter Darmkrebs
Monotherapie
zeigt die Nebenwirkungen, die bei ≥ 5 % der Patienten aus der Zusammenfassung der beiden Phase-3-Studien bei metastasiertem Darmkrebs der ersten Wahl auftraten. Insgesamt 596 Patienten mit metastasiertem Kolorektalkarzinom wurden mit 1250 mg/m2 XELODA zweimal täglich über 2 Wochen behandelt, gefolgt von einer einwöchigen Ruhephase, und 593 Patienten erhielten 5-FU und Leucovorin im Mayo-Schema (20 mg/m2 Leucovorin IV, gefolgt von 425 mg/m2 IV Bolus 5-FU, an den Tagen 1-5, alle 28 Tage). In der gepoolten kolorektalen Datenbank betrug die mediane Behandlungsdauer 139 Tage bei den mit Capecitabin behandelten Patienten und 140 Tage bei den mit 5-FU/LV behandelten Patienten. Insgesamt 78 (13 %) bzw. 63 (11 %) der mit Capecitabin bzw. 5-FU/LV behandelten Patienten brachen die Behandlung aufgrund von Nebenwirkungen/zwischenzeitlichen Erkrankungen ab. Insgesamt 82 Todesfälle aufgrund aller Ursachen traten entweder während der Studie oder innerhalb von 28 Tagen nach Erhalt des Studienmedikaments auf: 50 (8,4 %) Patienten wurden randomisiert zu XELODA 500 mg und 32 (5,4 %) randomisiert zu 5-FU/LV.
Brustkrebs
In Kombination mit Docetaxel
Die folgenden Daten werden für die Kombinationsstudie mit XELODA 500 mg und Docetaxel bei Patientinnen mit metastasiertem Brustkrebs in und gezeigt. Im Kombinationsarm mit XELODA und Docetaxel bestand die Behandlung aus oral verabreichtem XELODA 1250 mg/m2 zweimal täglich als intermittierende Therapie (2 Wochen Behandlung gefolgt von 1 Woche ohne Behandlung) für mindestens 6 Wochen und Docetaxel wurde als 1-stündige intravenöse Infusion verabreicht eine Dosis von 75 mg/m2 am ersten Tag jedes 3-Wochen-Zyklus für mindestens 6 Wochen. Im Monotherapie-Arm wurde Docetaxel als 1-stündige intravenöse Infusion in einer Dosis von 100 mg/m2 am ersten Tag jedes 3-wöchigen Zyklus für mindestens 6 Wochen verabreicht. Die mittlere Behandlungsdauer betrug 129 Tage im Kombinationsarm und 98 Tage im Monotherapiearm. Insgesamt 66 Patienten (26 %) im Kombinationsarm und 49 (19 %) im Monotherapiearm brachen die Studie wegen Nebenwirkungen ab. Der Prozentsatz der Patienten, die aufgrund von Nebenwirkungen eine Dosisreduktion benötigten, betrug 65 % im Kombinationsarm und 36 % im Monotherapiearm. Der Prozentsatz der Patienten, die aufgrund von Nebenwirkungen eine Behandlungsunterbrechung benötigten, betrug im Kombinationsarm 79 %. Behandlungsunterbrechungen waren Teil des Dosisanpassungsplans für den Kombinationstherapiearm, nicht jedoch für die mit Docetaxel-Monotherapie behandelten Patienten.
Monotherapie
Die folgenden Daten werden für die Studie an Brustkrebspatientinnen im Stadium IV gezeigt, die 2 Wochen lang zweimal täglich eine Dosis von 1250 mg/m2 erhielten, gefolgt von einer 1-wöchigen Ruhephase. Die mittlere Behandlungsdauer betrug 114 Tage. Insgesamt 13 von 162 Patienten (8 %) brachen die Behandlung aufgrund von Nebenwirkungen/interkurrenten Erkrankungen ab.
Klinisch relevante Nebenwirkungen bei
Klinisch relevante Nebenwirkungen, die bei
Monotherapie (metastasierter Darmkrebs, adjuvanter Darmkrebs, metastasierter Brustkrebs)
Magen-Darm: Blähungen, Dysphagie, Proktalgie, Aszites (0,1 %), Magengeschwür (0,1 %), Ileus (0,3 %), toxische Darmerweiterung, Gastroenteritis (0,1 %)
Haut & Unterhaut.: Nagelerkrankung (0,1 %), vermehrtes Schwitzen (0,1 %), Lichtempfindlichkeitsreaktion (0,1 %), Hautgeschwüre, Pruritus, Radiatio-Recall-Syndrom (0,2 %)
Allgemein: Brustschmerzen (0,2 %), grippeähnliche Erkrankung, Hitzewallungen, Schmerzen (0,1 %), Heiserkeit, Reizbarkeit, Schwierigkeiten beim Gehen, Durst, Brustraumforderung, Kollaps, Fibrose (0,1 %), Blutung, Ödem, Sedierung
Neurologisch: Schlaflosigkeit, Ataxie (0,5 %), Tremor, Dysphasie, Enzephalopathie (0,1 %), Koordinationsstörungen, Dysarthrie, Bewusstlosigkeit (0,2 %), Gleichgewichtsstörungen
Stoffwechsel: Gewichtszunahme, Kachexie (0,4 %), Hypertriglyzeridämie (0,1 %), Hypokaliämie, Hypomagnesiämie
Auge: Bindehautentzündung
Atmung: Husten (0,1 %), Epistaxis (0,1 %), Asthma (0,2 %), Hämoptyse, Atemnot (0,1 %), Dyspnoe
Herz: Tachykardie (0,1 %), Bradykardie, Vorhofflimmern, ventrikuläre Extrasystolen, Extrasystolen, Myokarditis (0,1 %), Perikarderguss
Infektionen: Laryngitis (1,0 %), Bronchitis (0,2 %), Lungenentzündung (0,2 %), Bronchopneumonie (0,2 %), Keratokonjunktivitis, Sepsis (0,3 %), Pilzinfektionen (einschließlich Candidose) (0,2 %)
Bewegungsapparat: Myalgie, Knochenschmerzen (0,1 %), Arthritis (0,1 %), Muskelschwäche
Blut & Lymphe: Leukopenie (0,2 %), Gerinnungsstörung (0,1 %), Knochenmarkdepression (0,1 %), idiopathische Thrombozytopenie purpura (1,0 %), Panzytopenie (0,1 %)
Gefäß: Hypotonie (0,2 %), Hypertonie (0,1 %), Lymphödem (0,1 %), Lungenembolie (0,2 %), Schlaganfall (0,1 %)
Psychiatrie: Depression, Verwirrtheit (0,1 %)
Nieren: Nierenfunktionsstörung (0,6 %)
Ohr: Schwindel
Hepatobiliär: Leberfibrose (0,1 %), Hepatitis (0,1 %), cholestatische Hepatitis (0,1 %), abnorme Leberfunktionstests
Immunsystem: Arzneimittelüberempfindlichkeit (0,1 %)
XELODA 500 mg in Kombination mit Docetaxel (metastasierender Brustkrebs)
Magen-Darm: Ileus (0,4 %), nekrotisierende Enterokolitis (0,4 %), Ösophagusulkus (0,4 %), hämorrhagischer Durchfall (0,8 %)
Neurologisch: Ataxie (0,4 %), Synkope (1,2 %), Geschmacksverlust (0,8 %), Polyneuropathie (0,4 %), Migräne (0,4 %)
Herz: supraventrikuläre Tachykardie (0,4 %)
Infektion: neutropenische Sepsis (2,4 %), Sepsis (0,4 %), Bronchopneumonie (0,4 %) Blut &
Lymphatisch: Agranulozytose (0,4 %), Prothrombin erniedrigt (0,4 %)
Gefäß: Hypotonie (1,2 %), venöse Phlebitis und Thrombophlebitis (0,4 %), orthostatische Hypotonie (0,8 %)
Nieren: Nierenversagen (0,4 %)
Hepatobiliär: Gelbsucht (0,4 %), anormale Leberfunktionstests (0,4 %), Leberversagen (0,4 %), hepatisches Koma (0,4 %), Hepatotoxizität (0,4 %)
Immunsystem: Überempfindlichkeit (1,2 %)
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden nach der Markteinführung beobachtet: Angioödem, Leberversagen, Tränenwegsstenose, akutes Nierenversagen infolge von Dehydratation, einschließlich tödlichem Ausgang [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ], kutaner Lupus erythematodes, Hornhauterkrankungen einschließlich Keratitis, toxische Leukenzephalopathie, schwere Hautreaktionen wie Stevens-Johnson-Syndrom und toxische epidermale Nekrolyse (TEN) [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN kann ein anhaltendes oder schweres Hand-Fuß-Syndrom schließlich zum Verlust von Fingerabdrücken führen [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]
Bei Kontakt mit zerkleinerten XELODA 500 mg Tabletten wurden die folgenden Nebenwirkungen berichtet: Augenreizung und -schwellung, Hautausschlag, Durchfall, Parästhesien, Kopfschmerzen, Magenreizung, Erbrechen und Übelkeit.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Arzneimittelwechselwirkungen
Antikoagulanzien
Veränderte Gerinnungsparameter und/oder Blutungen wurden bei Patienten berichtet, die XELODA 500 mg gleichzeitig mit von Cumarin abgeleiteten Antikoagulanzien wie Warfarin und Phenprocoumon einnahmen [siehe KASTENWARNUNG ]. Diese Ereignisse traten innerhalb von mehreren Tagen und bis zu mehreren Monaten nach Beginn der Behandlung mit XELODA 500 mg und in einigen Fällen innerhalb von 1 Monat nach Beendigung der Behandlung mit XELODA auf. Diese Ereignisse traten bei Patienten mit und ohne Lebermetastasen auf. In einer Wechselwirkungsstudie mit einer Einzeldosis Warfarin gab es einen signifikanten Anstieg der mittleren AUC von S-Warfarin [siehe KLINISCHE PHARMAKOLOGIE ]. Der maximal beobachtete INR-Wert stieg um 91 %. Diese Wechselwirkung ist wahrscheinlich auf eine Hemmung von Cytochrom P450 2C9 durch Capecitabin und/oder seine Metaboliten zurückzuführen.
Phenytoin
Der Phenytoinspiegel sollte bei Patienten, die XELODA 500 mg einnehmen, sorgfältig überwacht werden, und die Phenytoindosis muss möglicherweise reduziert werden [siehe DOSIERUNG UND ANWENDUNG ]. Postmarketing-Berichte weisen darauf hin, dass einige Patienten, die XELODA 500 mg und Phenytoin erhielten, Toxizitäten im Zusammenhang mit erhöhten Phenytoinspiegeln aufwiesen. Es wurden keine formellen Arzneimittelwechselwirkungsstudien mit Phenytoin durchgeführt, aber der Wechselwirkungsmechanismus ist vermutlich die Hemmung des CYP2C9-Isoenzyms durch Capecitabin und/oder seine Metaboliten.
Leucovorin
Die Konzentration von 5-Fluorouracil wird erhöht und seine Toxizität kann durch Leucovorin verstärkt werden. Bei älteren Patienten, die wöchentlich Leucovorin und Fluorouracil erhielten, wurde über Todesfälle durch schwere Enterokolitis, Durchfall und Dehydration berichtet.
CYP2C9-Substrate
Abgesehen von Warfarin wurden keine formellen Arzneimittelwechselwirkungsstudien zwischen XELODA und anderen CYP2C9-Substraten durchgeführt. Vorsicht ist geboten, wenn XELODA zusammen mit CYP2C9-Substraten verabreicht wird.
Allopurinol
Die gleichzeitige Anwendung mit Allopurinol kann die Konzentration der aktiven Metaboliten von Capecitabin verringern [siehe KLINISCHE PHARMAKOLOGIE ], was die Wirksamkeit von XELODA verringern kann. Vermeiden Sie die Anwendung von Allopurinol während der Behandlung mit XELODA.
Arzneimittel-Lebensmittel-Wechselwirkung
Es wurde gezeigt, dass Nahrung sowohl die Rate als auch das Ausmaß der Resorption von Capecitabin verringert [siehe KLINISCHE PHARMAKOLOGIE ]. In allen klinischen Studien wurden die Patienten angewiesen, XELODA 500 mg innerhalb von 30 Minuten nach einer Mahlzeit zu verabreichen. Es wird empfohlen, XELODA zusammen mit Nahrung einzunehmen [siehe DOSIERUNG UND ANWENDUNG ].
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Koagulopathie
Bei Patienten, die gleichzeitig mit Capecitabin und oralen Cumarin-Derivaten als Antikoagulanzien behandelt werden, sollte das Ansprechen auf Antikoagulanzien (INR oder Prothrombinzeit) engmaschig und häufig überwacht werden, und die Antikoagulanziendosis sollte entsprechend angepasst werden [siehe Abschnitt 4.4]. KASTENWARNUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Durchfall
XELODA 500 mg kann manchmal schweren Durchfall hervorrufen. Patienten mit schwerem Durchfall sollten sorgfältig überwacht und bei Dehydrierung mit Flüssigkeit und Elektrolyten ersetzt werden. Bei 875 Patienten mit entweder metastasiertem Brust- oder Darmkrebs, die eine XELODA-Monotherapie erhielten, betrug die mediane Zeit bis zum ersten Auftreten von Diarrhoe Grad 2 bis 4 34 Tage (Bereich von 1 bis 369 Tage). Die mediane Dauer von Diarrhoe Grad 3 bis 4 betrug 5 Tage. National Cancer Institute of Canada (NCIC) Grad 2 Diarrhoe ist definiert als eine Zunahme von 4 bis 6 Stuhlgängen/Tag oder nächtlicher Stuhlgang, Grad 3 Diarrhoe als eine Zunahme von 7 bis 9 Stuhlgängen/Tag oder Inkontinenz und Malabsorption und Grad 4 Diarrhoe als eine Zunahme von ≥ 10 Stuhlgängen/Tag oder stark blutiger Durchfall oder die Notwendigkeit einer parenteralen Unterstützung. Wenn Diarrhö Grad 2, 3 oder 4 auftritt, sollte die Verabreichung von XELODA sofort unterbrochen werden, bis die Diarrhoe abgeklungen ist oder an Intensität auf Grad 1 abgenommen hat [siehe DOSIERUNG UND ANWENDUNG ]. Standardbehandlungen gegen Durchfall (z. B. Loperamid) werden empfohlen.
Nekrotisierende Enterokolitis (Typhlitis) wurde berichtet.
Kardiotoxizität
Die bei XELODA 500 mg beobachtete Kardiotoxizität umfasst Myokardinfarkt/Ischämie, Angina pectoris, Rhythmusstörungen, Herzstillstand, Herzinsuffizienz, plötzlichen Tod, elektrokardiographische Veränderungen und Kardiomyopathie. Diese Nebenwirkungen können häufiger bei Patienten mit koronarer Herzkrankheit in der Vorgeschichte auftreten.
Dihydropyrimidin-Dehydrogenase-Mangel
Basierend auf Postmarketing-Berichten haben Patienten mit bestimmten homozygoten oder bestimmten zusammengesetzten heterozygoten Mutationen im DPD-Gen, die zu einem vollständigen oder nahezu vollständigen Fehlen der DPD-Aktivität führen, ein erhöhtes Risiko für einen akuten frühen Beginn einer Toxizität und schwere, lebensbedrohliche oder tödliche Nebenwirkungen durch XELODA verursachte Reaktionen (z. B. Mukositis, Durchfall, Neutropenie und Neurotoxizität). Patienten mit partieller DPD-Aktivität können auch ein erhöhtes Risiko für schwere, lebensbedrohliche oder tödliche Nebenwirkungen haben, die durch XELODA verursacht werden.
Unterbrechen oder dauerhaftes Absetzen von XELODA 500 mg basierend auf der klinischen Beurteilung des Beginns, der Dauer und des Schweregrads der beobachteten Toxizitäten bei Patienten mit Anzeichen einer akuten, früh einsetzenden oder ungewöhnlich schweren Toxizität, die auf ein nahezu vollständiges oder vollständiges Fehlen einer DPD-Aktivität hindeuten kann. Keine XELODA-Dosis hat sich für Patienten mit vollständig fehlender DPD-Aktivität als sicher erwiesen. Es liegen keine ausreichenden Daten vor, um eine bestimmte Dosis bei Patienten mit partieller DPD-Aktivität, gemessen mit einem bestimmten Test, zu empfehlen.
Dehydration und Nierenversagen
Dehydration wurde beobachtet und kann akutes Nierenversagen verursachen, das tödlich sein kann. Patienten mit vorbestehender eingeschränkter Nierenfunktion oder die gleichzeitig XELODA mit bekannten nephrotoxischen Wirkstoffen erhalten, sind einem höheren Risiko ausgesetzt. Patienten mit Anorexie, Asthenie, Übelkeit, Erbrechen oder Durchfall können schnell dehydrieren. Überwachen Sie die Patienten, wenn XELODA 500 mg verabreicht wird, um eine anfängliche Dehydration zu verhindern und zu korrigieren. Wenn eine Dehydratation Grad 2 (oder höher) auftritt, sollte die Behandlung mit XELODA sofort unterbrochen und die Dehydratation korrigiert werden. Die Behandlung sollte nicht wieder aufgenommen werden, bis der Patient rehydriert ist und alle auslösenden Ursachen behoben oder kontrolliert wurden. Je nach Bedarf sollten Dosisanpassungen für das auslösende unerwünschte Ereignis vorgenommen werden [siehe DOSIERUNG UND ANWENDUNG ].
Patienten mit mäßig eingeschränkter Nierenfunktion zu Studienbeginn benötigen eine Dosisreduktion [siehe DOSIERUNG UND ANWENDUNG ]. Patienten mit leichter und mittelschwerer Nierenfunktionsstörung zu Studienbeginn sollten sorgfältig auf Nebenwirkungen überwacht werden. Eine sofortige Unterbrechung der Therapie mit anschließender Dosisanpassung wird empfohlen, wenn ein Patient ein unerwünschtes Ereignis 2. bis 4. Grades entwickelt, wie in [siehe DOSIERUNG UND ANWENDUNG , Verwendung in bestimmten Bevölkerungsgruppen , und KLINISCHE PHARMAKOLOGIE ].
Embryo-fötale Toxizität
Basierend auf Ergebnissen aus Reproduktionsstudien an Tieren und seinem Wirkungsmechanismus kann XELODA den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird [siehe KLINISCHE PHARMAKOLOGIE ]. Begrenzt verfügbare Daten reichen nicht aus, um die Anwendung von XELODA 500 mg bei Schwangeren zu informieren. In Reproduktionsstudien an Tieren verursachte die Verabreichung von Capecitabin an trächtige Tiere während der Organogenese bei Patienten, die die empfohlene Dosis erhielten, Embryoletalität und Teratogenität bei Mäusen und Embryoletalität bei Affen, wenn die Exposition (AUC) dem 0,2- bzw. 0,6-Fachen entsprach [siehe Verwendung in bestimmten Bevölkerungsgruppen ]. Informieren Sie schwangere Frauen über das potenzielle Risiko für einen Fötus. Weisen Sie Frauen im gebärfähigen Alter an, während der Behandlung und für 6 Monate nach der letzten XELODA-Dosis eine wirksame Verhütungsmethode anzuwenden [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Mukokutane und dermatologische Toxizität
Bei Patienten, die mit XELODA behandelt werden, können schwere Schleimhautreaktionen, einige mit tödlichem Ausgang, wie Stevens-Johnson-Syndrom und toxische epidermale Nekrolyse (TEN) auftreten [siehe NEBENWIRKUNGEN ]. XELODA 500 mg sollte bei Patienten, bei denen eine schwere mukokutane Reaktion auftritt, die möglicherweise auf die Behandlung mit XELODA zurückzuführen ist, dauerhaft abgesetzt werden.
Das Hand-Fuß-Syndrom (palmar-plantare Erythrodysästhesie oder Chemotherapie-induziertes akrales Erythem) ist eine kutane Toxizität. Die mediane Zeit bis zum Auftreten betrug 79 Tage (Bereich von 11 bis 360 Tagen) mit einem Schweregrad von Grad 1 bis 3 bei Patienten, die XELODA 500 mg als Monotherapie im metastasierten Setting erhielten. Grad 1 ist gekennzeichnet durch eines der folgenden Merkmale: Taubheitsgefühl, Dysästhesie/Parästhesie, Kribbeln, schmerzlose Schwellung oder Erythem der Hände und/oder Füße und/oder Beschwerden, die normale Aktivitäten nicht stören. Das Hand-Fuß-Syndrom Grad 2 ist definiert als schmerzhaftes Erythem und Schwellung der Hände und/oder Füße und/oder Beschwerden, die die Aktivitäten des Patienten im täglichen Leben beeinträchtigen. Das Hand-Fuß-Syndrom Grad 3 ist definiert als feuchte Abschuppung, Ulzeration, Blasenbildung oder starke Schmerzen der Hände und/oder Füße und/oder starke Beschwerden, die dazu führen, dass der Patient nicht in der Lage ist, zu arbeiten oder Aktivitäten des täglichen Lebens auszuführen. Ein anhaltendes oder schweres Hand-Fuß-Syndrom (Grad 2 und höher) kann schließlich zum Verlust von Fingerabdrücken führen, was die Patientenidentifikation beeinträchtigen könnte. Wenn ein Hand-Fuß-Syndrom Grad 2 oder 3 auftritt, sollte die Verabreichung von XELODA unterbrochen werden, bis das Ereignis abgeklungen ist oder die Intensität auf Grad 1 abnimmt. Nach einem Hand-Fuß-Syndrom Grad 3 sollten die nachfolgenden Dosen von XELODA 500 mg verringert werden [ sehen DOSIERUNG UND ANWENDUNG ].
Hyperbilirubinämie
Bei 875 Patienten mit entweder metastasiertem Brust- oder Darmkrebs, die mindestens eine Dosis XELODA 1250 mg/m2 zweimal täglich als Monotherapie über 2 Wochen gefolgt von einer einwöchigen Ruhephase erhielten, trat eine Hyperbilirubinämie Grad 3 (1,5-3 x ULN) auf 15,2 % (n = 133) der Patienten und Hyperbilirubinämie Grad 4 (> 3 x ULN) trat bei 3,9 % (n = 34) der Patienten auf. Von 566 Patienten mit Lebermetastasen zu Studienbeginn und 309 Patienten ohne Lebermetastasen zu Studienbeginn trat bei 22,8 % bzw. 12,3 % eine Hyperbilirubinämie Grad 3 oder 4 auf. Von den 167 Patienten mit Hyperbilirubinämie Grad 3 oder 4 hatten 18,6 % (n = 31) auch postbaseline Erhöhungen (Grad 1 bis 4, ohne Erhöhungen zu Studienbeginn) der alkalischen Phosphatase und 27,5 % (n = 46) hatten postbaseline Erhöhungen der Transaminasen bei jederzeit (nicht notwendigerweise gleichzeitig). Die Mehrheit dieser Patienten, 64,5 % (n = 20) und 71,7 % (n = 33), hatte zu Studienbeginn Lebermetastasen. Darüber hinaus hatten 57,5 % (n = 96) und 35,3 % (n = 59) der 167 Patienten Erhöhungen (Grad 1 bis 4) sowohl vor als auch nach dem Ausgangswert der alkalischen Phosphatase bzw. der Transaminasen. Nur 7,8 % (n = 13) und 3,0 % (n = 5) hatten Erhöhungen der alkalischen Phosphatase oder der Transaminasen Grad 3 oder 4.
Bei den 596 Patienten, die mit XELODA 500 mg als Erstlinientherapie für metastasierten Darmkrebs behandelt wurden, war die Inzidenz von Hyperbilirubinämie Grad 3 oder 4 ähnlich der Gesamtsicherheitsdatenbank aus klinischen Studien zur XELODA 500 mg-Monotherapie. Die mediane Zeit bis zum Auftreten einer Hyperbilirubinämie 3. oder 4. Grades in der Darmkrebspopulation betrug 64 Tage, und das mediane Gesamtbilirubin stieg von 8 μm/l zu Studienbeginn auf 13 μm/l während der Behandlung mit XELODA. Von den 136 Darmkrebspatienten mit Hyperbilirubinämie Grad 3 oder 4 hatten 49 Patienten eine Hyperbilirubinämie Grad 3 oder 4 als letzten gemessenen Wert, von denen 46 zu Studienbeginn Lebermetastasen aufwiesen.
Bei 251 Patientinnen mit metastasiertem Brustkrebs, die eine Kombination aus XELODA 500 mg und Docetaxel erhielten, trat bei 7 % (n = 17) eine Hyperbilirubinämie Grad 3 (1,5 bis 3 x ULN) und bei 2 % eine Hyperbilirubinämie Grad 4 (> 3 x ULN) auf. (n = 5).
Wenn arzneimittelbedingte Bilirubinerhöhungen von Grad 3 bis 4 auftreten, sollte die Verabreichung von XELODA 500 mg sofort unterbrochen werden, bis die Hyperbilirubinämie auf ≤ 3,0 x ULN abgefallen ist [siehe empfohlene Dosisanpassungen unten DOSIERUNG UND ANWENDUNG ].
Hämatologisch
Von 875 Patienten mit entweder metastasiertem Brust- oder Darmkrebs, die eine Dosis von 1250 mg/m2 zweimal täglich als Monotherapie über 2 Wochen erhielten, gefolgt von einer einwöchigen Ruhephase, hatten 3,2 %, 1,7 % und 2,4 % der Patienten Grad 3 oder 4 Neutropenie, Thrombozytopenie bzw. Abfall des Hämoglobins. Von 251 Patientinnen mit metastasiertem Brustkrebs, die eine Dosis XELODA in Kombination mit Docetaxel erhielten, hatten 68 % eine Neutropenie Grad 3 oder 4, 2,8 % eine Thrombozytopenie Grad 3 oder 4 und 9,6 % eine Anämie Grad 3 oder 4.
Patienten mit einer Neutrophilenzahl von
Geriatrische Patienten
Bei Patienten im Alter von ≥ 80 Jahren kann es häufiger zu Nebenwirkungen vom Grad 3 oder 4 kommen. Von 875 Patienten mit entweder metastasiertem Brust- oder Darmkrebs, die XELODA 500 mg als Monotherapie erhielten, trat bei 62 % der 21 mit XELODA 500 mg behandelten Patienten im Alter von ≥ 80 Jahren ein behandlungsbedingtes unerwünschtes Ereignis 3. oder 4. Grades auf: Durchfall bei 6 (28,6 %) , Übelkeit bei 3 (14,3 %), Hand-Fuß-Syndrom bei 3 (14,3 %) und Erbrechen bei 2 (9,5 %) Patienten. Von den 10 Patienten im Alter von 70 Jahren und älter (kein Patient war älter als 80 Jahre), die mit XELODA 500 mg in Kombination mit Docetaxel behandelt wurden, erlitten 30 % (3 von 10) der Patienten Diarrhoe und Stomatitis Grad 3 oder 4 und 40 % (4 von 10) hatten ein Hand-Fuß-Syndrom Grad 3.
Bei den 67 Patienten im Alter von ≥ 60 Jahren, die XELODA in Kombination mit Docetaxel erhielten, traten behandlungsbedingte Nebenwirkungen Grad 3 oder 4, schwerwiegende behandlungsbedingte Nebenwirkungen, Behandlungsabbrüche aufgrund von Nebenwirkungen, Behandlungsabbrüche aufgrund von Nebenwirkungen und Behandlung auf Behandlungsabbrüche innerhalb der ersten beiden Behandlungszyklen war höher als in der Patientengruppe
Von 995 Patienten, die XELODA als adjuvante Therapie bei Dukes-C-Darmkrebs nach Resektion des Primärtumors erhielten, trat bei 41 % der 398 mit XELODA behandelten Patienten im Alter von ≥ 65 Jahren ein behandlungsbedingtes unerwünschtes Ereignis 3. oder 4. Grades auf: Hand-und -Fußsyndrom bei 75 (18,8 %), Durchfall bei 52 (13,1 %), Stomatitis bei 12 (3,0 %), Neutropenie/Granulozytopenie bei 11 (2,8 %), Erbrechen bei 6 (1,5 %) und Übelkeit bei 5 (1,3 %) Patienten. Bei Patienten im Alter von ≥ 65 Jahren (alle randomisierten Populationen; Capecitabin 188 Patienten, 5-FU/LV 208 Patienten), die wegen eines Dukes-C-Kolonkarzinoms nach Resektion des Primärtumors behandelt wurden, wurden die Hazard Ratios für das krankheitsfreie Überleben und das Gesamtüberleben für XELODA im Vergleich zu 5-FU/LV waren 1,01 (95 % KI 0,80 – 1,27) bzw. 1,04 (95 % KI 0,79 – 1,37).
Leberinsuffizienz
Patienten mit leichter bis mittelschwerer Leberfunktionsstörung aufgrund von Lebermetastasen sollten während der Verabreichung von XELODA sorgfältig überwacht werden. Die Auswirkung einer schweren Leberfunktionsstörung auf die Verfügbarkeit von XELODA ist nicht bekannt [siehe Verwendung in bestimmten Bevölkerungsgruppen und KLINISCHE PHARMAKOLOGIE ].
Kombination mit anderen Drogen
Die Anwendung von XELODA in Kombination mit Irinotecan wurde nicht ausreichend untersucht.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( INFORMATIONEN ZUM PATIENTEN ).
Durchfall
Informieren Sie Patienten mit Diarrhoe Grad 2 (eine Zunahme von 4 bis 6 Stuhlgängen/Tag oder nächtlicher Stuhlgang) oder mehr oder mit schwerem blutigem Durchfall mit starken Bauchschmerzen und Fieber, die Einnahme von XELODA abzubrechen. Informieren Sie die Patienten über die Anwendung von Antidiarrhoika (z. B. Loperamid) zur Behandlung von Durchfall [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Kardiotoxizität
Weisen Sie die Patienten auf das Risiko einer Kardiotoxizität hin und weisen Sie sie darauf hin, dass sie bei neu auftretenden Brustschmerzen, Kurzatmigkeit, Schwindel oder Benommenheit sofort ihren Arzt kontaktieren oder eine Notaufnahme aufsuchen sollen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Dihydropyrimidin-Dehydrogenase-Mangel
Raten Sie den Patienten, ihren Arzt zu benachrichtigen, wenn sie einen bekannten DPD-Mangel haben. Weisen Sie Patienten darauf hin, wenn bei ihnen eine vollständige oder nahezu vollständige Abwesenheit von DPD-Aktivität vorliegt, da sie einem erhöhten Risiko einer akuten frühen Toxizität und schweren, lebensbedrohlichen oder tödlichen Nebenwirkungen ausgesetzt sind, die durch XELODA verursacht werden (z. B. Mukositis, Durchfall, Neutropenie und Neurotoxizität) [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Dehydration und Nierenversagen
Weisen Sie Patienten mit Dehydration Grad 2 oder höher an (IV-Flüssigkeiten angegeben WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Wichtige Verwaltungsanweisungen
Empfehlen Sie den Patienten, XELODA 500 mg Tabletten unzerkaut mit Wasser innerhalb von 30 Minuten nach einer Mahlzeit zu schlucken. Weisen Sie Patienten und Pflegekräfte darauf hin, XELODA-Tabletten nicht zu zerdrücken oder zu schneiden. Raten Sie Patienten, wenn sie XELODA Tabletten nicht ganz schlucken können, ihren Arzt zu informieren [siehe DOSIERUNG UND ANWENDUNG ].
Überempfindlichkeit und Angioödem
Weisen Sie die Patienten darauf hin, dass XELODA schwere Überempfindlichkeitsreaktionen und Angioödeme verursachen kann. Weisen Sie Patienten mit bekannter Überempfindlichkeit gegenüber Capecitabin oder 5-Fluorouracil an, ihren Arzt zu informieren [siehe KONTRAINDIKATIONEN ]. Weisen Sie Patienten, die Überempfindlichkeitsreaktionen oder mukokutane Symptome entwickeln (z. B. Urtikaria, Hautausschlag, Erythem, Juckreiz oder Schwellungen von Gesicht, Lippen, Zunge oder Rachen, die das Schlucken oder Atmen erschweren), an, die Einnahme von XELODA abzubrechen und unverzüglich ihren Arzt zu kontaktieren oder in eine Notaufnahme gehen. [sehen NEBENWIRKUNGEN ].
Brechreiz
Weisen Sie Patienten mit Übelkeit 2. Grades (Nahrungsaufnahme deutlich verringert, aber in der Lage, mit Unterbrechungen zu essen) oder höher an, die Einnahme von XELODA 500 mg sofort abzubrechen und sich zur Behandlung der Übelkeit an ihren Arzt zu wenden [siehe NEBENWIRKUNGEN ].

Erbrechen
Weisen Sie Patienten mit Erbrechen Grad 2 (2 bis 5 Episoden innerhalb von 24 Stunden) oder mehr an, die Einnahme von XELODA sofort abzubrechen und sich zur Behandlung des Erbrechens an ihren Arzt zu wenden [siehe NEBENWIRKUNGEN ].
Hand-Fuß-Syndrom
Weisen Sie Patienten mit einem Hand-Fuß-Syndrom Grad 2 (schmerzhaftes Erythem und Schwellung der Hände und/oder Füße und/oder Beschwerden, die die Aktivitäten des Patienten beeinträchtigen) oder größer an, die Einnahme von XELODA 500 mg sofort abzubrechen und sich an Ihren Arzt zu wenden . Informieren Sie die Patienten darüber, dass die Einleitung einer symptomatischen Behandlung empfohlen wird und das Hand-Fuß-Syndrom zum Verlust von Fingerabdrücken führen kann, was die persönliche Identifizierung beeinträchtigen könnte [siehe NEBENWIRKUNGEN ].
Stomatitis
Informieren Sie Patienten mit Stomatitis Grad 2 (schmerzhaftes Erythem, Ödem oder Geschwüre im Mund oder an der Zunge, aber in der Lage zu essen) oder höher, die Einnahme von XELODA sofort abzubrechen und sich an Ihren Arzt zu wenden [siehe NEBENWIRKUNGEN ].
Fieber und Neutropenie
Informieren Sie Patienten, die ein Fieber von 100,5 °F oder mehr oder andere Anzeichen einer möglichen Infektion entwickeln, sich an ihren Arzt zu wenden [siehe NEBENWIRKUNGEN ].
Embryo-fötale Toxizität
Weisen Sie gebärfähige Frauen auf das potenzielle Risiko für einen Fötus hin und weisen Sie sie darauf hin, während der Behandlung mit XELODA 500 mg und für 6 Monate nach der letzten Dosis eine wirksame Empfängnisverhütung anzuwenden. Raten Sie Frauen, ihren Arzt über eine bekannte oder vermutete Schwangerschaft zu informieren [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , Verwendung in bestimmten Bevölkerungsgruppen ].
Weisen Sie männliche Patienten mit gebärfähigen Partnerinnen an, während der Behandlung mit XELODA 500 mg und für 3 Monate nach der letzten Dosis eine wirksame Empfängnisverhütung anzuwenden [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Stillzeit
Raten Sie Frauen, während der Behandlung mit XELODA 500 mg und für 2 Wochen nach der letzten Dosis nicht zu stillen [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Es wurden keine angemessenen Studien zum kanzerogenen Potenzial von Capecitabin durchgeführt. Capecitabin war in vitro nicht mutagen gegenüber Bakterien (Ames-Test) oder Säugerzellen (V79/HPRT-Genmutationstest des chinesischen Hamsters). Capecitabin war in vitro klastogen gegenüber humanen peripheren Blutlymphozyten, aber nicht klastogen in vivo gegenüber Mausknochenmark (Mikrokerntest). Fluorouracil verursacht Mutationen in Bakterien und Hefen. Fluorouracil verursacht auch Chromosomenanomalien im Maus-Mikrokerntest in vivo.
In Studien zur Fertilität und allgemeinen Reproduktionsleistung bei weiblichen Mäusen störten orale Capecitabin-Dosen von 760 mg/kg/Tag (ca. 2300 mg/m2/Tag) die Brunst und verursachten folglich eine Abnahme der Fertilität. Bei Mäusen, die schwanger wurden, überlebten keine Föten diese Dosis. Die Brunststörung war reversibel. Bei Männern verursachte diese Dosis degenerative Veränderungen in den Hoden, einschließlich einer Abnahme der Anzahl von Spermatozyten und Spermatiden. In separaten pharmakokinetischen Studien führte diese Dosis bei Mäusen zu 5'-DFUR-AUC-Werten, die etwa das 0,7-Fache der entsprechenden Werte bei Patienten betrugen, denen die empfohlene Tagesdosis verabreicht wurde.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Zusammenfassung der Risiken
Basierend auf Ergebnissen aus Reproduktionsstudien an Tieren und seinem Wirkungsmechanismus kann XELODA 500 mg den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird [siehe KLINISCHE PHARMAKOLOGIE ]. Begrenzt verfügbare Humandaten reichen nicht aus, um über das arzneimittelbedingte Risiko während der Schwangerschaft zu informieren. In Reproduktionsstudien an Tieren führte die Verabreichung von Capecitabin an trächtige Tiere während der Organogenese zu Embryoletalität und Teratogenität bei Mäusen und Embryoletalität bei Affen bei 0,2- bzw. 0,6-facher Exposition (AUC) bei Patienten, die die empfohlene Dosis erhielten [siehe Daten ]. Informieren Sie schwangere Frauen über das potenzielle Risiko für einen Fötus.
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten für die angegebene Population ist unbekannt. In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwere Geburtsfehler und Fehlgeburten bei klinisch anerkannten Schwangerschaften bei 2–4 % bzw. 15–20 %.
Daten
Tierdaten
Die orale Verabreichung von Capecitabin an trächtige Mäuse während der Organogenese in einer Dosis von 198 mg/kg/Tag verursachte Missbildungen und Embryoletalität. In separaten pharmakokinetischen Studien führte diese Dosis bei Mäusen zu 5'-DFUR-AUC-Werten, die etwa dem 0,2-fachen der AUC-Werte bei Patienten entsprachen, denen die empfohlene Tagesdosis verabreicht wurde. Fehlbildungen bei Mäusen umfassten Gaumenspalten, Anophthalmie, Mikrophthalmie, Oligodaktylie, Polydaktylie, Syndaktylie, Knickschwanz und Erweiterung der Hirnventrikel. Die orale Verabreichung von Capecitabin an trächtige Affen während der Organogenese in einer Dosis von 90 mg/kg/Tag verursachte fötale Letalität. Diese Dosis führte zu 5'-DFUR-AUC-Werten, die etwa dem 0,6-fachen der AUC-Werte bei Patienten entsprachen, denen die empfohlene Tagesdosis verabreicht wurde.
Stillzeit
Zusammenfassung der Risiken
Es liegen keine Informationen zum Vorhandensein von Capecitabin in der Muttermilch oder zu seinen Auswirkungen auf die Milchproduktion oder den gestillten Säugling vor. Capecitabin-Metabolite waren in der Milch von laktierenden Mäusen vorhanden [siehe Daten ]. Aufgrund der Möglichkeit schwerwiegender Nebenwirkungen durch Capecitabin-Exposition bei gestillten Säuglingen ist Frauen anzuraten, während der Behandlung mit XELODA 500 mg und für 2 Wochen nach der letzten Dosis nicht zu stillen.
Daten
Laktierende Mäuse, denen eine orale Einzeldosis Capecitabin verabreicht wurde, schieden signifikante Mengen von Capecitabin-Metaboliten in die Milch aus.
Weibchen und Männchen mit reproduktivem Potenzial
Schwangerschaftstest
Bei Frauen im gebärfähigen Alter wird vor Beginn der Behandlung mit XELODA ein Schwangerschaftstest empfohlen.
Empfängnisverhütung
Frauen
XELODA 500 mg kann den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird [siehe Schwangerschaft ]. Raten Sie Frauen im gebärfähigen Alter, während der Behandlung und für 6 Monate nach der letzten XELODA-Dosis eine wirksame Verhütungsmethode anzuwenden.
Männchen
Auf der Grundlage von Befunden zur genetischen Toxizität sollten männliche Patienten mit gebärfähigen Partnerinnen angewiesen werden, während der Behandlung und für 3 Monate nach der letzten XELODA-Dosis eine wirksame Empfängnisverhütung anzuwenden [siehe Nichtklinische Toxikologie ].
Unfruchtbarkeit
Basierend auf Tierversuchen kann XELODA die Fertilität bei weiblichen und männlichen Fortpflanzungsfähigen beeinträchtigen [siehe Nichtklinische Toxikologie ].
Pädiatrische Verwendung
Die Sicherheit und Wirksamkeit von XELODA bei pädiatrischen Patienten wurde nicht nachgewiesen. In zwei einarmigen Studien an pädiatrischen Patienten mit neu diagnostizierten Hirnstammgliomen und hochgradigen Gliomen wurde kein klinischer Nutzen nachgewiesen. In beiden Studien erhielten pädiatrische Patienten eine pädiatrische Prüfformulierung von Capecitabin gleichzeitig mit und nach Abschluss der Strahlentherapie (Gesamtdosis von 5580 cGy in 180 cGy-Fraktionen). Die relative Bioverfügbarkeit der Prüfformulierung war ähnlich wie bei XELODA 500 mg.
Die erste Studie wurde an 22 pädiatrischen Patienten (Durchschnittsalter 8 Jahre, Bereich 5–17 Jahre) mit neu diagnostizierten, nicht disseminierten intrinsischen diffusen Hirnstammgliomen und hochgradigen Gliomen durchgeführt. Im Dosisfindungsteil der Studie erhielten die Patienten Capecitabin mit begleitender Strahlentherapie in Dosen von 500 mg/m2 bis 850 mg/m2 alle 12 Stunden für bis zu 9 Wochen. Nach einer 2-wöchigen Pause erhielten die Patienten 1250 mg/m2 Capecitabin alle 12 Stunden an den Tagen 1-14 eines 21-Tage-Zyklus für bis zu 3 Zyklen. Die maximal tolerierte Dosis (MTD) von Capecitabin, die gleichzeitig mit einer Strahlentherapie verabreicht wurde, betrug 650 mg/m2 alle 12 Stunden. Die wichtigsten dosislimitierenden Toxizitäten waren palmar-plantare Erythrodysästhesie und Erhöhung der Alanin-Aminotransferase (ALT).
Die zweite Studie wurde mit 34 weiteren pädiatrischen Patienten mit neu diagnostizierten, nicht disseminierten intrinsischen diffusen Hirnstammgliomen (Durchschnittsalter 7 Jahre, Bereich 3–16 Jahre) und 10 pädiatrischen Patienten durchgeführt, die die MTD von Capecitabin in der Dosisfindungsstudie erhielten und erfüllten die Zulassungskriterien für diese Studie. Alle Patienten erhielten 650 mg/m2 Capecitabin alle 12 Stunden mit begleitender Strahlentherapie für bis zu 9 Wochen. Nach einer 2-wöchigen Pause erhielten die Patienten 1250 mg/m2 Capecitabin alle 12 Stunden an den Tagen 1-14 eines 21-Tage-Zyklus für bis zu 3 Zyklen.
Bei pädiatrischen Patienten mit neu diagnostizierten intrinsischen Hirnstammgliomen, die Capecitabin erhielten, war im Vergleich zu einer ähnlichen Population von pädiatrischen Patienten, die an anderen klinischen Studien teilnahmen, keine Verbesserung der progressionsfreien Überlebensrate nach einem Jahr und der Gesamtüberlebensrate nach einem Jahr zu verzeichnen.
Das Nebenwirkungsprofil von Capecitabin stimmte mit dem bekannten Nebenwirkungsprofil bei Erwachsenen überein, mit Ausnahme von Laboranomalien, die häufiger bei pädiatrischen Patienten auftraten. Die am häufigsten berichteten Laboranomalien (Inzidenz pro Patient ≥ 40 %) waren erhöhte ALT (75 %), Lymphozytopenie (73 %), Leukopenie (73 %), Hypokaliämie (68 %), Thrombozytopenie (57 %), Hypoalbuminämie (55 %), Neutropenie (50 %), niedriger Hämatokrit (50 %), Hypokalzämie (48 %), Hypophosphatämie (45 %) und Hyponatriämie (45 %).
Geriatrische Verwendung
Ärzte sollten besonders darauf achten, die Nebenwirkungen von XELODA 500 mg bei älteren Patienten zu überwachen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Leberinsuffizienz
Seien Sie vorsichtig, wenn Patienten mit leichter bis mittelschwerer Leberfunktionsstörung aufgrund von Lebermetastasen mit XELODA behandelt werden. Die Auswirkung einer schweren Leberfunktionsstörung auf XELODA ist nicht bekannt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und KLINISCHE PHARMAKOLOGIE ].
Niereninsuffizienz
Patienten mit mäßiger (Kreatinin-Clearance = 30 bis 50 ml/min) und schwerer (Kreatinin-Clearance KONTRAINDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , DOSIERUNG UND ANWENDUNG , und KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Zu den Manifestationen einer akuten Überdosierung gehören Übelkeit, Erbrechen, Durchfall, Magen-Darm-Reizung und -Blutungen sowie Knochenmarkdepression. Die medizinische Behandlung einer Überdosierung sollte übliche unterstützende medizinische Interventionen umfassen, die darauf abzielen, die vorliegenden klinischen Manifestationen zu korrigieren. Obwohl keine klinischen Erfahrungen mit der Dialyse zur Behandlung einer Überdosierung von XELODA 500 mg vorliegen, kann die Dialyse bei der Reduzierung der zirkulierenden Konzentrationen von 5'-DFUR, einem niedermolekularen Metaboliten der Muttersubstanz, von Vorteil sein.
Einzeldosen von XELODA 500 mg waren für Mäuse, Ratten und Affen in Dosen von bis zu 2000 mg/kg (das 2,4-, 4,8- und 9,6-fache der empfohlenen Tagesdosis beim Menschen auf mg/m2-Basis) nicht tödlich.
KONTRAINDIKATIONEN
Schwere Nierenfunktionsstörung
XELODA ist kontraindiziert bei Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance unter 30 ml/min [Cockroft und Gault]) [vgl Verwendung in bestimmten Bevölkerungsgruppen und KLINISCHE PHARMAKOLOGIE ].
Überempfindlichkeit
XELODA 500 mg ist bei Patienten mit bekannter Überempfindlichkeit gegen Capecitabin oder einen seiner Bestandteile kontraindiziert. XELODA ist bei Patienten mit bekannter Überempfindlichkeit gegen 5-Fluorouracil kontraindiziert.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Enzyme wandeln Capecitabin in vivo in 5-Fluorouracil (5-FU) um. Sowohl normale als auch Tumorzellen metabolisieren 5-FU zu 5-Fluor-2'-desoxyuridinmonophosphat (FdUMP) und 5-Fluoruridintriphosphat (FUTP). Diese Metaboliten verursachen Zellschädigungen durch zwei verschiedene Mechanismen. Erstens binden FdUMP und der Folat-Cofaktor N5-10-Methylentetrahydrofolat an Thymidylat-Synthase (TS), um einen kovalent gebundenen ternären Komplex zu bilden. Diese Bindung hemmt die Bildung von Thymidylat aus 2'-Desoxyuridylat. Thymidylat ist die notwendige Vorstufe des für die DNA-Synthese essentiellen Thymidintriphosphats, so dass ein Mangel an dieser Verbindung die Zellteilung hemmen kann. Zweitens können nukleäre Transkriptionsenzyme während der Synthese von RNA fälschlicherweise FUTP anstelle von Uridintriphosphat (UTP) einbauen. Dieser Stoffwechselfehler kann die RNA-Verarbeitung und die Proteinsynthese stören.
Pharmakokinetik
Absorption
Nach oraler Gabe von 1255 mg/m2 BID an Krebspatienten erreichte Capecitabin innerhalb von etwa 1,5 Stunden (Tmax) maximale Blutspiegel, wobei die maximalen 5-FU-Spiegel etwas später, nach 2 Stunden, auftraten. Nahrung reduzierte sowohl die Rate als auch das Ausmaß der Resorption von Capecitabin, wobei die mittlere Cmax und AUC0-∞ um 60 % bzw. 35 % abnahmen. Cmax und AUC0-∞ von 5-FU wurden ebenfalls durch Nahrung um 43 % bzw. 21 % reduziert. Nahrung verzögerte Tmax von beiden Elternteilen und 5-FU um 1,5 Stunden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , DOSIERUNG UND ANWENDUNG , und Arzneimittel-Lebensmittel-Wechselwirkung ].
Die Pharmakokinetik von XELODA 500 mg und seinen Metaboliten wurde bei etwa 200 Krebspatienten in einem Dosierungsbereich von 500 bis 3500 mg/m2/Tag untersucht. Über diesen Bereich hinweg war die Pharmakokinetik von XELODA und seinem Metaboliten 5'-DFCR dosisproportional und veränderte sich im Laufe der Zeit nicht. Die Anstiege der AUCs von 5'-DFUR und 5-FU waren jedoch größer als proportional zur Erhöhung der Dosis, und die AUC von 5-FU war an Tag 14 um 34 % höher als an Tag 1. Die interindividuelle Variabilität in der Cmax und AUC von 5-FU waren größer als 85 %.
Verteilung
Die Plasmaproteinbindung von Capecitabin und seinen Metaboliten beträgt weniger als 60 % und ist nicht konzentrationsabhängig. Capecitabin wurde hauptsächlich an Humanalbumin gebunden (ca. 35 %). XELODA 500 mg hat ein geringes Potenzial für pharmakokinetische Wechselwirkungen im Zusammenhang mit der Plasmaproteinbindung.
Bioaktivierung und Stoffwechsel
Capecitabin wird weitgehend enzymatisch zu 5-FU metabolisiert. In der Leber hydrolysiert eine 60-kDa-Carboxylesterase einen Großteil der Verbindung zu 5'-Desoxy-5-Fluorcytidin (5'-DFCR). Cytidin-Desaminase, ein Enzym, das in den meisten Geweben, einschließlich Tumoren, vorkommt, wandelt anschließend 5'-DFCR in 5'-DFUR um. Das Enzym Thymidin-Phosphorylase (dThdPase) hydrolysiert dann 5'DFUR zum Wirkstoff 5-FU. Viele Gewebe im ganzen Körper exprimieren Thymidinphosphorylase. Einige menschliche Karzinome exprimieren dieses Enzym in höheren Konzentrationen als das umgebende normale Gewebe. Nach oraler Verabreichung von XELODA 7 Tage vor der Operation bei Patienten mit kolorektalem Karzinom betrug das mediane Verhältnis der 5-FU-Konzentration in kolorektalen Tumoren zu angrenzenden Geweben 2,9 (Bereich von 0,9 bis 8,0). Diese Verhältnisse wurden bei Brustkrebspatientinnen nicht bewertet oder mit einer 5-FU-Infusion verglichen.
Stoffwechselweg von Capecitabin zu 5-FU
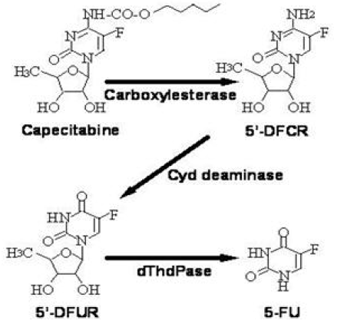
Das Enzym Dihydropyrimidin-Dehydrogenase hydriert 5-FU, das Stoffwechselprodukt von Capecitabin, zu dem viel weniger toxischen 5-Fluor-5,6-Dihydro-Fluorouracil (FUH2). Dihydropyrimidinase spaltet den Pyrimidinring, um 5-Fluor-ureido-propionsäure (FUPA) zu ergeben. Schließlich spaltet β-Ureido-Propionase FUPA zu α-Fluor-β-Alanin (FBAL), das im Urin ausgeschieden wird.
Enzymatische In-vitro-Studien mit menschlichen Lebermikrosomen zeigten, dass Capecitabin und seine Metaboliten (5'-DFUR, 5'-DFCR, 5-FU und FBAL) den Metabolismus von Testsubstraten durch die Cytochrom-P450-Isoenzyme 1A2, 2A6, 3A4, 2C19, 2D6 und 2E1.
Ausscheidung
Capecitabin und seine Metaboliten werden überwiegend im Urin ausgeschieden; 95,5 % der verabreichten Capecitabin-Dosis werden im Urin wiedergefunden. Die fäkale Ausscheidung ist minimal (2,6 %). Der Hauptmetabolit, der im Urin ausgeschieden wird, ist FBAL, das 57 % der verabreichten Dosis ausmacht. Etwa 3 % der verabreichten Dosis werden unverändert im Urin ausgeschieden. Die Eliminationshalbwertszeit sowohl der Ausgangssubstanz Capecitabin als auch von 5-FU betrug etwa 0,75 Stunden.
Einfluss von Alter, Geschlecht und Rasse auf die Pharmakokinetik von Capecitabin
Eine Populationsanalyse der gepoolten Daten aus den beiden großen kontrollierten Studien bei Patienten mit metastasiertem Darmkrebs (n = 505), denen XELODA 500 mg mit 1250 mg/m2 zweimal täglich verabreicht wurde, zeigte, dass Geschlecht (202 Frauen und 303 Männer) und Rasse (455 weiße/kaukasische Patienten, 22 schwarze Patienten und 28 Patienten anderer Rasse) haben keinen Einfluss auf die Pharmakokinetik von 5'DFUR, 5-FU und FBAL. Das Alter hat keinen signifikanten Einfluss auf die Pharmakokinetik von 5'-DFUR und 5-FU im Bereich von 27 bis 86 Jahren. Eine 20-prozentige Zunahme des Alters führt zu einer 15-prozentigen Zunahme der AUC von FBAL [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN und DOSIERUNG UND ANWENDUNG ].
Nach oraler Verabreichung von 825 mg/m2 Capecitabin zweimal täglich über 14 Tage hatten japanische Patienten (n = 18) eine etwa 36 % niedrigere Cmax und eine 24 % niedrigere AUC für Capecitabin als die kaukasischen Patienten (n = 22). Japanische Patienten hatten auch eine etwa 25 % niedrigere Cmax und eine 34 % niedrigere AUC für FBAL als die kaukasischen Patienten. Die klinische Bedeutung dieser Unterschiede ist nicht bekannt. Bei der Exposition gegenüber anderen Metaboliten (5'-DFCR, 5'-DFUR und 5FU) traten keine signifikanten Unterschiede auf.
Auswirkung einer Leberinsuffizienz
XELODA wurde bei 13 Patienten mit leichter bis mittelschwerer Leberfunktionsstörung aufgrund von Lebermetastasen, definiert durch einen zusammengesetzten Score aus Bilirubin, AST/ALT und alkalischer Phosphatase, nach einer Einzeldosis von 1255 mg/m2 XELODA untersucht. Sowohl AUC0-∞ als auch Cmax von Capecitabin stiegen bei Patienten mit Leberfunktionsstörung um 60 % im Vergleich zu Patienten mit normaler Leberfunktion (n = 14). Die AUC0-∞ und Cmax von 5-FU wurden nicht beeinflusst. Bei Patienten mit leichter bis mittelschwerer Leberfunktionsstörung aufgrund von Lebermetastasen ist bei der Verabreichung von XELODA 500 mg Vorsicht geboten. Die Auswirkung einer schweren Leberfunktionsstörung auf XELODA 500 mg ist nicht bekannt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in speziellen Bevölkerungsgruppen ].
Auswirkung einer Niereninsuffizienz
Nach oraler Gabe von 1250 mg/m2 Capecitabin zweimal täglich an Krebspatienten mit unterschiedlich stark eingeschränkter Nierenfunktion zeigten Patienten mit mäßiger (Kreatinin-Clearance = 30 bis 50 ml/min) und schwerer (Kreatinin-Clearance 80 ml/min). Die systemische Exposition gegenüber 5'-DFUR war bei Patienten mit mäßiger bzw. schwerer Nierenfunktionsstörung um 42 % bzw. 71 % höher als bei normalen Patienten. Die systemische Capecitabin-Exposition war sowohl bei Patienten mit mäßiger als auch mit schwerer Nierenfunktionsstörung um etwa 25 % höher [siehe DOSIERUNG UND ANWENDUNG , KONTRAINDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , und Verwendung in speziellen Bevölkerungsgruppen ].
Wirkung von Capecitabin auf die Pharmakokinetik von Warfarin
Bei vier Krebspatienten erhöhte die chronische Gabe von Capecitabin (1250 mg/m2 zweimal täglich) mit einer Einzeldosis von 20 mg Warfarin die mittlere AUC von S-Warfarin um 57 % und verringerte seine Clearance um 37 %. Die zu Studienbeginn korrigierte INR-AUC stieg bei diesen 4 Patienten um das 2,8-Fache, und der maximal beobachtete mittlere INR-Wert war um 91 % erhöht [siehe KASTENWARNUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Wirkung von Antazida auf die Pharmakokinetik von Capecitabin
Wenn Maalox® (20 ml), ein Aluminiumhydroxid- und Magnesiumhydroxid-haltiges Antazidum, unmittelbar nach XELODA (1250 mg/m2, n = 12 Krebspatienten) verabreicht wurde, erhöhten sich AUC und Cmax um 16 % bzw. 35 %. für Capecitabin und um 18 % bzw. 22 % für 5'-DFCR. Bei den anderen drei Hauptmetaboliten (5'-DFUR, 5-FU, FBAL) von XELODA wurde keine Wirkung beobachtet.
Wirkung von Allopurinol auf Capecitabin
In der veröffentlichten Literatur wurde berichtet, dass die gleichzeitige Anwendung mit Allopurinol die Umwandlung von Capecitabin in die aktiven Metaboliten FdUMP und FUTP verringern kann; die klinische Bedeutung wurde jedoch nicht vollständig charakterisiert.
Wirkung von Capecitabin auf die Pharmakokinetik von Docetaxel und umgekehrt
Eine Phase-1-Studie untersuchte die Wirkung von XELODA auf die Pharmakokinetik von Docetaxel (Taxotere®) und die Wirkung von Docetaxel auf die Pharmakokinetik von XELODA wurde bei 26 Patienten mit soliden Tumoren durchgeführt. XELODA hat keinen Einfluss auf die Pharmakokinetik von Docetaxel (Cmax und AUC) und Docetaxel hat keinen Einfluss auf die Pharmakokinetik von Capecitabin und dem 5-FU-Vorläufer 5'-DFUR.
Klinische Studien
Adjuvans Darmkrebs
Eine multizentrische, randomisierte, kontrollierte klinische Studie der Phase 3 bei Patienten mit Dickdarmkrebs des Typs Dukes C (X-ACT) lieferte Daten zur Verwendung von XELODA zur adjuvanten Behandlung von Patienten mit Dickdarmkrebs. Das primäre Ziel der Studie war der Vergleich des krankheitsfreien Überlebens (DFS) bei Patienten, die XELODA erhielten, mit denen, die nur IV 5-FU/LV erhielten. In dieser Studie wurden 1987 Patienten randomisiert entweder einer Behandlung mit XELODA 1250 mg/m2 oral zweimal täglich für 2 Wochen, gefolgt von einer einwöchigen Ruhephase, gegeben als 3-Wochen-Zyklen für insgesamt 8 Zyklen (24 Wochen) oder IV Bolus 5-FU 425 mg/m2 und 20 mg/m2 IV Leucovorin an den Tagen 1 bis 5, gegeben als 4-Wochen-Zyklen für insgesamt 6 Zyklen (24 Wochen). Die Patienten in der Studie mussten zwischen 18 und 75 Jahre alt sein und einen histologisch bestätigten Dickdarmkrebs im Stadium C nach Dukes mit mindestens einem positiven Lymphknoten aufweisen und sich (innerhalb von 8 Wochen vor der Randomisierung) einer vollständigen Resektion des Primärtumors unterzogen haben ohne makroskopischen oder mikroskopischen Nachweis eines verbleibenden Tumors. Die Patienten mussten außerdem keine vorherige zytotoxische Chemotherapie oder Immuntherapie (außer Steroide) erhalten haben und einen ECOG-Leistungsstatus von 0 oder 1 (KPS ≥ 70 %), ANC ≥ 1,5 x 109/l, Thrombozyten ≥ 100 x 109/l, Serumkreatinin ≤ haben 1,5 ULN, Gesamtbilirubin ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN und CEA innerhalb der normalen Grenzen zum Zeitpunkt der Randomisierung.
Die demografischen Ausgangsdaten für XELODA- und 5-FU/LV-Patienten sind in Tabelle 10 dargestellt. Die Ausgangscharakteristika waren zwischen den Armen gut ausgewogen.
Alle Patienten mit normaler Nierenfunktion oder leichter Nierenfunktionsstörung begannen die Behandlung mit der vollen Anfangsdosis von 1250 mg/m2 oral zweimal täglich. Bei Patienten mit mäßig eingeschränkter Nierenfunktion (berechnete Kreatinin-Clearance 30 bis 50 ml/min) zu Studienbeginn wurde die Anfangsdosis reduziert [vgl DOSIERUNG UND ANWENDUNG ]. Anschließend wurden bei allen Patienten die Dosen bei Bedarf entsprechend der Toxizität angepasst. Das Dosismanagement für XELODA 500 mg umfasste Dosisreduktionen, Zyklusverzögerungen und Behandlungsunterbrechungen (siehe Tabelle 11).
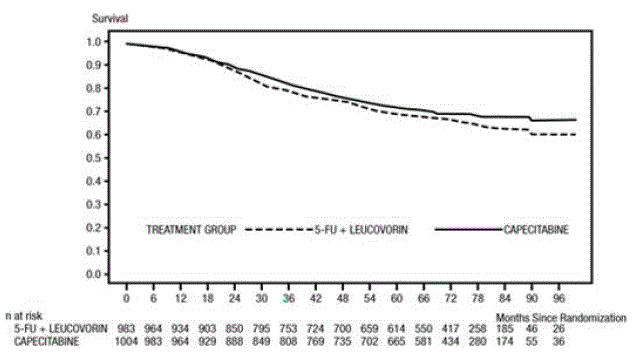
Das mediane Follow-up zum Zeitpunkt der Analyse betrug 83 Monate (6,9 Jahre). Die Hazard Ratio für DFS für XELODA 500 mg im Vergleich zu 5-FU/LV betrug 0,88 (95 % KI 0,77 – 1,01) (siehe und Abbildung 1 ). Da die obere zweiseitige 95 %-Konfidenzgrenze der Hazard Ratio unter 1,20 lag, war XELODA 5-FU/LV nicht unterlegen. Die Wahl der Nichtunterlegenheitsspanne von 1,20 entspricht dem Erhalt von etwa 75 % der 5-FU/LV-Wirkung auf das DFS. Die Hazard Ratio für XELODA im Vergleich zu 5-FU/LV in Bezug auf das Gesamtüberleben betrug 0,86 (95 % KI 0,74 – 1,01). Die 5-Jahres-Gesamtüberlebensraten betrugen 71,4 % für XELODA und 68,4 % für 5-FU/LV (siehe Abbildung 2).
Abbildung 1 Kaplan-Meier-Schätzungen des krankheitsfreien Überlebens (alle randomisierten Populationen)a
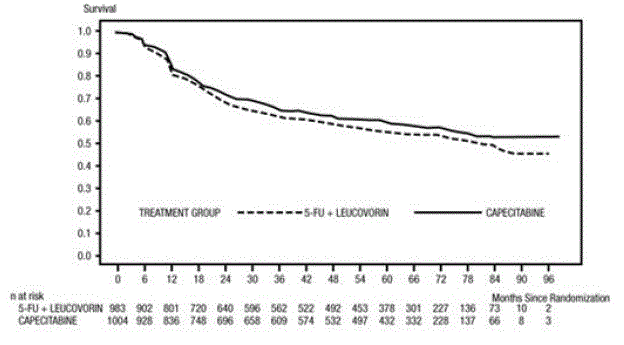
Abbildung 2 Kaplan-Meier-Schätzungen des Gesamtüberlebens (alle randomisierten Populationen)
Metastasierter Darmkrebs
Allgemein
Die empfohlene Dosis von XELODA 500 mg wurde in einer unverblindeten, randomisierten klinischen Studie bestimmt, in der die Wirksamkeit und Sicherheit einer kontinuierlichen Therapie mit Capecitabin (1331 mg/m2/Tag in zwei getrennten Dosen, n = 39), einer intermittierenden Therapie mit Capecitabin ( 2510 mg/m2/Tag in zwei aufgeteilten Dosen, n=34) und intermittierende Therapie mit Capecitabin in Kombination mit oralem Leucovorin (LV) (Capecitabin 1657 mg/m2/Tag in zwei aufgeteilten Dosen, n=35; Leucovorin 60 mg/ Tag) bei Patienten mit fortgeschrittenem und/oder metastasiertem kolorektalen Karzinom in der Erstlinientherapie mit Metastasen. Es gab keinen offensichtlichen Vorteil in Bezug auf die Ansprechrate bei der Zugabe von Leucovorin zu XELODA; die Toxizität war jedoch erhöht. XELODA, 1250 mg/m2 zweimal täglich für 14 Tage, gefolgt von einer einwöchigen Pause, wurde für die weitere klinische Entwicklung ausgewählt, basierend auf dem allgemeinen Sicherheits- und Wirksamkeitsprofil der drei untersuchten Schemata.
Monotherapie
Daten aus zwei offenen, multizentrischen, randomisierten, kontrollierten klinischen Studien mit 1207 Patienten unterstützen die Anwendung von XELODA in der Erstlinienbehandlung von Patienten mit metastasiertem kolorektalen Karzinom. Die beiden klinischen Studien waren im Design identisch und wurden in 120 Zentren in verschiedenen Ländern durchgeführt. Studie 1 wurde in den USA, Kanada, Mexiko und Brasilien durchgeführt; Studie 2 wurde in Europa, Israel, Australien, Neuseeland und Taiwan durchgeführt. Insgesamt wurden in beiden Studien 603 Patienten für die Behandlung mit XELODA 500 mg in einer Dosis von 1250 mg/m2 zweimal täglich für 2 Wochen, gefolgt von einer einwöchigen Ruhephase, randomisiert und in dreiwöchigen Zyklen verabreicht; 604 Patienten wurden randomisiert einer Behandlung mit 5-FU und Leucovorin (20 mg/m2 Leucovorin i.v., gefolgt von 425 mg/m2 i.v. Bolus 5-FU, an den Tagen 1 bis 5, alle 28 Tage) zugeteilt.
In beiden Studien wurden das Gesamtüberleben, die Zeit bis zur Progression und die Ansprechrate (vollständiges und partielles Ansprechen) bewertet. Die Reaktionen wurden anhand der Kriterien der Weltgesundheitsorganisation definiert und einem verblindeten unabhängigen Prüfungsausschuss (IRC) vorgelegt. Unterschiede in den Bewertungen zwischen dem Prüfarzt und dem IRC wurden vom Sponsor, verblindet für den Behandlungsarm, gemäß einem festgelegten Algorithmus ausgeglichen. Das Überleben wurde basierend auf einer Nichtunterlegenheitsanalyse bewertet.
Die demografischen Daten zu Studienbeginn für XELODA- und 5-FU/LV-Patienten sind in Tabelle 13 dargestellt.
Die Wirksamkeitsendpunkte für die beiden Phase-3-Studien sind in und dargestellt
Abbildung 3 Kaplan-Meier-Kurve für das Gesamtüberleben gepoolter Daten (Studien 1 und 2)
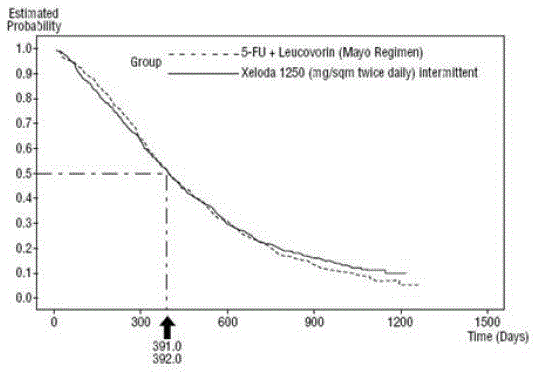
XELODA war 5-FU/LV in Bezug auf die objektive Ansprechrate in Studie 1 und Studie 2 überlegen. Die Ähnlichkeit von XELODA 500 mg und 5-FU/LV in diesen Studien wurde durch Untersuchung des potenziellen Unterschieds zwischen den beiden Behandlungen bewertet. Um sicherzustellen, dass XELODA 500 mg einen klinisch bedeutsamen Überlebenseffekt hat, wurden statistische Analysen durchgeführt, um den Prozentsatz des Überlebenseffekts von 5-FU/LV zu bestimmen, der von XELODA beibehalten wurde. Die Schätzung des Überlebenseffekts von 5-FU/LV wurde aus einer Metaanalyse von zehn randomisierten Studien aus der veröffentlichten Literatur abgeleitet, in denen 5-FU mit Regimen von 5-FU/LV verglichen wurden, die den in diesen Studien verwendeten Kontrollarmen ähnlich waren 1 und 2. Die Methode zum Vergleich der Behandlungen bestand darin, den schlimmsten Fall (95 % Konfidenzobergrenze) auf den Unterschied zwischen 5-FU/LV und XELODA 500 mg zu untersuchen und zu zeigen, dass ein Verlust von mehr als 50 % des 5- FU/LV-Überlebenseffekt wurde ausgeschlossen. Es wurde gezeigt, dass der Prozentsatz des Überlebenseffekts von 5-FU/LV bei mindestens 61 % für Studie 2 und 10 % für Studie 1 aufrechterhalten wurde. Das gepoolte Ergebnis stimmt mit einer Beibehaltung von mindestens 50 % des Effekts von 5 überein -FU/LV. Es sollte beachtet werden, dass diese Werte für die erhaltene Wirkung auf der Obergrenze der Differenz zwischen 5-FU/LV und XELODA 500 mg basieren. Diese Ergebnisse schließen die Möglichkeit einer echten Äquivalenz von XELODA 500 mg zu 5-FU/LV nicht aus (siehe und Figur 3 ).
Brustkrebs
XELODA wurde in klinischen Studien in Kombination mit Docetaxel (Taxotere®) und als Monotherapie untersucht.
In Kombination mit Docetaxel
Die Dosis von XELODA, die in der klinischen Phase-3-Studie in Kombination mit Docetaxel verwendet wurde, basierte auf den Ergebnissen einer Phase-1-Studie, in der eine Reihe von Dosen von Docetaxel in 3-wöchigen Zyklen in Kombination mit einem intermittierenden Regime von XELODA (14 Tage der Behandlung, gefolgt von einer 7-tägigen Ruhephase) wurden ausgewertet. Das Kombinationsdosierungsschema wurde basierend auf dem Verträglichkeitsprofil von 75 mg/m2 Docetaxel, verabreicht in 3-wöchigen Zyklen, in Kombination mit 1250 mg/m2 zweimal täglich für 14 Tage von XELODA 500 mg, verabreicht in 3-wöchigen Zyklen, ausgewählt. Die zugelassene Dosis von 100 mg/m2 Docetaxel, verabreicht in 3-Wochen-Zyklen, war der Kontrollarm der Phase-3-Studie.
XELODA in Kombination mit Docetaxel wurde in einer unverblindeten, multizentrischen, randomisierten Studie in 75 Zentren in Europa, Nordamerika, Südamerika, Asien und Australien untersucht. Insgesamt 511 Patientinnen mit metastasiertem Brustkrebs, die resistent gegen oder während oder nach einer anthrazyklinhaltigen Therapie wiederauftraten oder während oder innerhalb von 2 Jahren nach Abschluss einer anthrazyklinhaltigen adjuvanten Therapie rezidivierten, wurden eingeschlossen. Zweihundertfünfundfünfzig (255) Patienten wurden randomisiert und erhielten XELODA 1250 mg/m2 zweimal täglich für 14 Tage, gefolgt von 1 Woche ohne Behandlung, und Docetaxel 75 mg/m2 als einstündige intravenöse Infusion, verabreicht in 3-wöchigen Zyklen. Im Monotherapie-Arm erhielten 256 Patienten Docetaxel 100 mg/m2 als einstündige intravenöse Infusion, verabreicht in 3-wöchigen Zyklen. Demografische Daten der Patienten sind in Tabelle 16 angegeben.
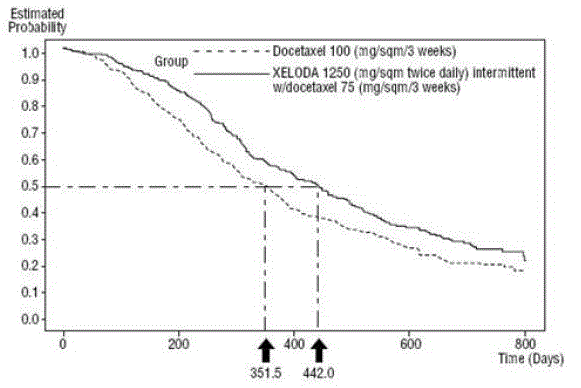
XELODA in Kombination mit Docetaxel führte zu einer statistisch signifikanten Verbesserung der Zeit bis zum Fortschreiten der Krankheit, des Gesamtüberlebens und der objektiven Ansprechrate im Vergleich zur Monotherapie mit Docetaxel, wie in und gezeigt Abbildung 5 .
Abbildung 4 Kaplan-Meier-Schätzungen für die Zeit bis zum Fortschreiten der Krankheit XELODA und Docetaxel vs. Docetaxel
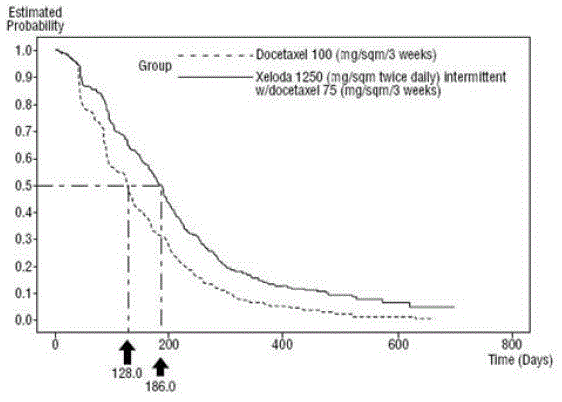
Abbildung 5 Kaplan-Meier-Schätzungen zum Überleben von XELODA und Docetaxel vs. Docetaxel
Monotherapie
Die Antitumoraktivität von XELODA als Monotherapie wurde in einer unverblindeten einarmigen Studie untersucht, die in 24 Zentren in den USA und Kanada durchgeführt wurde. Insgesamt wurden 162 Patientinnen mit Brustkrebs im Stadium IV aufgenommen. Der primäre Endpunkt war die Ansprechrate des Tumors bei Patienten mit messbarer Erkrankung, wobei das Ansprechen als Abnahme der Summe der Produkte der senkrechten Durchmesser der zweidimensional messbaren Erkrankung um ≥50 % für mindestens 1 Monat definiert ist. XELODA wurde in einer Dosis von 1255 mg/m2 zweimal täglich über 2 Wochen, gefolgt von einer 1-wöchigen Ruhephase, in dreiwöchigen Zyklen verabreicht. Die demografischen Ausgangsdaten und klinischen Charakteristika für alle Patienten (n=162) und diejenigen mit messbarer Erkrankung (n=135) sind in dargestellt. Resistenz wurde definiert als fortschreitende Erkrankung während der Behandlung, mit oder ohne anfängliches Ansprechen, oder Rückfall innerhalb von 6 Monaten nach Abschluss der Behandlung mit einem anthrazyklinhaltigen adjuvanten Chemotherapieschema.
Antitumorreaktionen bei Patienten mit einer Krankheit, die sowohl gegen Paclitaxel als auch gegen ein Anthracyclin resistent ist, sind in gezeigt
Für die Untergruppe von 43 Patienten, die doppelt resistent waren, betrug die mediane Zeit bis zur Progression 102 Tage und die mediane Überlebenszeit 255 Tage. Die objektive Ansprechrate in dieser Population wurde durch eine Ansprechrate von 18,5 % (1 CR, 24 PRs) in der Gesamtpopulation von 135 Patienten mit messbarer Erkrankung, die weniger resistent gegen eine Chemotherapie waren, gestützt (siehe ). Die mediane Zeit bis zur Progression betrug 90 Tage und die mediane Überlebenszeit 306 Tage.
INFORMATIONEN ZUM PATIENTEN
XELODA® (zeh-LOE-duh) (Capecitabin) Tabletten
Was sind die wichtigsten Informationen, die ich über XELODA 500 mg wissen sollte?
XELODA 500 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Sehen „Was sind die möglichen Nebenwirkungen von XELODA 500 mg?“ für weitere Informationen zu Nebenwirkungen.
Was ist XELODA?
XELODA ist ein verschreibungspflichtiges Arzneimittel zur Behandlung von Menschen mit:
Es ist nicht bekannt, ob XELODA 500 mg bei Kindern sicher und wirksam ist.
Nehmen Sie XELODA nicht ein, wenn Sie:
Sprechen Sie mit Ihrem Arzt, bevor Sie XELODA einnehmen, wenn Sie sich nicht sicher sind, ob Sie an einer der oben aufgeführten Erkrankungen leiden.
Informieren Sie vor der Einnahme von XELODA Ihren Arzt über alle Ihre Erkrankungen, auch wenn Sie:
Sehen „Was sind die wichtigsten Informationen, die ich über XELODA wissen sollte?“
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und pflanzlicher Nahrungsergänzungsmittel. XELODA kann die Wirkungsweise anderer Arzneimittel beeinflussen, und andere Arzneimittel können die Wirkungsweise von XELODA beeinflussen.
Informieren Sie sich über die Medikamente, die Sie einnehmen. Führen Sie eine Liste davon, um sie Ihrem Arzt und Apotheker zu zeigen, wenn Sie ein neues Arzneimittel erhalten.
Wie sollte ich XELODA 500 mg einnehmen?
Welche Nebenwirkungen kann XELODA haben?
XELODA kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Übelkeit und Erbrechen sind bei XELODA häufig. Wenn Sie Ihren Appetit verlieren, sich schwach fühlen und Übelkeit, Erbrechen oder Durchfall haben, können Sie schnell dehydrieren.
Brechen Sie die Einnahme von XELODA ab und wenden Sie sich sofort an Ihren Arzt, wenn Sie:
Wenn Ihre Anzahl weißer Blutkörperchen sehr niedrig ist, besteht ein erhöhtes Infektionsrisiko. Rufen Sie sofort Ihren Arzt an, wenn Sie Fieber von 100,5 °F oder mehr entwickeln oder andere Anzeichen und Symptome einer Infektion haben.
Bei Personen über 80 Jahren kann es wahrscheinlicher sein, dass unter XELODA schwere oder schwerwiegende Nebenwirkungen auftreten.
Zu den häufigsten Nebenwirkungen von XELODA 500 mg gehören:
Bei XELODA können schwere allergische Reaktionen auftreten. Informieren Sie Ihren Arzt, wenn Sie jemals eine allergische Reaktion auf Capecitabin oder 5-Fluorouracil hatten. Sehen „Nehmen Sie XELODA nicht ein, wenn Sie:“. Brechen Sie die Einnahme von XELODA 500 mg ab und wenden Sie sich sofort an Ihren Arzt oder suchen Sie eine Notaufnahme auf, wenn Sie eines der folgenden Symptome einer schweren allergischen Reaktion haben:
XELODA 500 mg kann bei Frauen und Männern Fruchtbarkeitsprobleme verursachen. Dies kann die Fähigkeit, ein Kind zu bekommen, beeinträchtigen. Sprechen Sie mit Ihrem Arzt, wenn Sie Bedenken hinsichtlich der Fruchtbarkeit haben.
Dies sind nicht alle möglichen Nebenwirkungen von XELODA.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist XELODA aufzubewahren?
Bewahren Sie XELODA und alle Arzneimittel außerhalb der Reichweite von Kindern auf.
Allgemeine Informationen zur sicheren und wirksamen Anwendung von XELODA.
Arzneimittel werden manchmal zu anderen als den in der Packungsbeilage aufgeführten Zwecken verschrieben. Verwenden Sie XELODA nicht für einen Zustand, für den es nicht verschrieben wurde. Geben Sie XELODA 500 mg nicht an andere Personen weiter, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden. Sie können Ihren Apotheker oder Gesundheitsdienstleister nach Informationen über XELODA 500 mg fragen, die für Angehörige der Gesundheitsberufe bestimmt sind.
Welche Inhaltsstoffe enthält XELODA 500 mg?
Wirkstoff: Capecitabin
Inaktive Zutaten: wasserfreie Lactose, Croscarmellose-Natrium, Hydroxypropylmethylcellulose, mikrokristalline Cellulose, Magnesiumstearat und gereinigtes Wasser. Die pfirsichfarbene oder hellpfirsichfarbene Filmbeschichtung enthält Hydroxypropylmethylcellulose, Talk, Titandioxid und synthetische gelbe und rote Eisenoxide.
Weitere Informationen finden Sie unter http://www.gene.com/patients/medicines/xeloda 500mg oder telefonisch unter 1-877-436-3683.
Diese Patienteninformation wurde von der US Food and Drug Administration genehmigt.