Rebetol 200mg Ribavirin Verwendung, Nebenwirkungen, Stärke und Dosierung. Preis in Online-Apotheke. Generika medikamente rezeptfrei.
Was ist Rebetol 200 mg und wie wird es angewendet?
Rebetol ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von chronischer Hepatitis C. Rebetol 200 mg kann allein oder zusammen mit anderen Medikamenten angewendet werden.
Rebetol gehört zu einer Klasse von Arzneimitteln, die Hepatitis-B-/Hepatitis-C-Mittel genannt werden; RSV-Agenten.
Es ist nicht bekannt, ob Rebetol 200 mg bei Kindern unter 3 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Rebetol haben?
Rebetol kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Rebetol. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
WARNUNG
RISIKO SCHWERER ERKRANKUNGEN UND RIBAVIRIN-ASSOZIIERTER WIRKUNGEN
BEZEICHNUNG
REBETOL (Ribavirin) ist ein synthetisches Nukleosid-Analogon (Purin-Analogon). Der chemische Name von Ribavirin ist 1-β-D-Ribofuranosyl-1H-1,2,4-triazol-3-carboxamid und hat die folgende Strukturformel (siehe Fig. 1).
Abbildung 1: Strukturformel
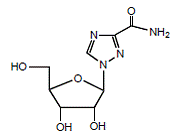
Ribavirin ist ein weißes, kristallines Pulver. Es ist leicht löslich in Wasser und leicht löslich in wasserfreiem Alkohol. Die Summenformel ist C8H12N4O5 und das Molekulargewicht ist 244,21.
REBETOL-Kapseln bestehen aus einem weißen Pulver in einer weißen, undurchsichtigen Gelatinekapsel. Jede Kapsel enthält 200 mg Ribavirin und die sonstigen Bestandteile mikrokristalline Cellulose, Lactose-Monohydrat, Croscarmellose-Natrium und Magnesiumstearat. Die Kapselhülle besteht aus Gelatine, Natriumlaurylsulfat, Siliziumdioxid und Titandioxid. Die Kapsel ist mit essbarer blauer pharmazeutischer Tinte bedruckt, die aus Schellack, wasserfreiem Ethylalkohol, Isopropylalkohol, n-Butylalkohol, Propylenglykol, Ammoniumhydroxid und FD&C Blue #2 Aluminiumlack hergestellt wird.
REBETOL 200 mg Lösung zum Einnehmen ist eine klare, farblose bis blass- oder hellgelbe Flüssigkeit mit Kaugummigeschmack. Jeder Milliliter der Lösung enthält 40 mg Ribavirin und die Hilfsstoffe Saccharose, Glycerin, Sorbitol, Propylenglycol, Natriumcitrat, Zitronensäure, Natriumbenzoat, natürliches und künstliches Aroma für Kaugummi #15864 und Wasser.
INDIKATIONEN
Chronische Hepatitis C (CHC)
REBETOL® (Ribavirin) in Kombination mit Interferon alfa-2b (pegyliert und nicht pegyliert) ist angezeigt für die Behandlung von chronischer Hepatitis C (CHC) bei Patienten ab 3 Jahren mit kompensierter Lebererkrankung [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , und Verwendung in bestimmten Populationen ].
Die folgenden Punkte sollten bei der Einleitung einer REBETOL-Kombinationstherapie mit PegIntron® oder INTRON A® berücksichtigt werden:
DOSIERUNG UND ANWENDUNG
REBETOL-Kapseln dürfen unter keinen Umständen geöffnet, zerdrückt oder zerbrochen werden. REBETOL 200 mg sollte mit Nahrung eingenommen werden [siehe KLINISCHE PHARMAKOLOGIE ]. REBETOL sollte nicht bei Patienten mit einer Kreatinin-Clearance von weniger als 50 ml/min angewendet werden.
REBETOL/PegIntron-Kombinationstherapie
Erwachsene Patienten
Die empfohlene Dosis von PegIntron beträgt 1,5 µg/kg/Woche subkutan in Kombination mit 800 bis 1400 mg REBETOL 200 mg Kapseln oral, basierend auf dem Körpergewicht des Patienten (siehe Tabelle 1). Das zu injizierende Volumen von PegIntron hängt von der Stärke von PegIntron und dem Körpergewicht des Patienten ab. Weitere Informationen zur Dosierung finden Sie auf dem Etikett von PegIntron.
Dauer der Behandlung – Interferon-Alpha-naive Patienten
Die Behandlungsdauer für Patienten mit Genotyp 1 beträgt 48 Wochen. Bei Patienten, die nach 12 Wochen keinen Abfall oder Verlust von HCV-RNA um mindestens 2 log10 erreichen oder wenn HCV-RNA nach 24 Wochen Therapie noch nachweisbar ist, sollte ein Absetzen der Therapie in Betracht gezogen werden. Patienten mit Genotyp 2 und 3 sollten 24 Wochen lang behandelt werden.
Behandlungsdauer – Wiederbehandlung mit PegIntron/REBETOL bei vorherigem Behandlungsversagen
Die Behandlungsdauer für Patienten mit vorherigem Therapieversagen beträgt 48 Wochen, unabhängig vom HCV-Genotyp. Bei erneut behandelten Patienten, die in Woche 12 der Therapie keine nicht nachweisbare HCV-RNA erreichen oder deren HCV-RNA nach 24 Wochen Therapie nachweisbar bleibt, ist es sehr unwahrscheinlich, dass sie eine SVR erreichen, und ein Absetzen der Therapie sollte in Betracht gezogen werden [siehe Klinische Studien ].
Pädiatrische Patienten
Die Dosierung für pädiatrische Patienten richtet sich bei PegIntron nach der Körperoberfläche und bei REBETOL nach dem Körpergewicht. Die empfohlene Dosis von PegIntron beträgt 60 mcg/m²/Woche subkutan in Kombination mit 15 mg/kg/Tag REBETOL 200 mg oral in zwei getrennten Dosen (siehe Tabelle 2) für pädiatrische Patienten im Alter von 3 bis 17 Jahren. Patienten, die während der Behandlung mit PegIntron/REBETOL 200 mg ihren 18. Geburtstag erreichen, sollten das pädiatrische Dosierungsschema beibehalten. Die Behandlungsdauer für Patienten mit Genotyp 1 beträgt 48 Wochen. Patienten mit Genotyp 2 und 3 sollten 24 Wochen lang behandelt werden.
REBETOL/INTRON Eine Kombinationstherapie
Erwachsene
Dauer der Behandlung – Interferon-Alpha-naive Patienten
Die empfohlene Dosis von INTRON A beträgt 3 Millionen IE dreimal wöchentlich subkutan. Die empfohlene Dosis von REBETOL-Kapseln hängt vom Körpergewicht des Patienten ab (siehe Tabelle 3). Die empfohlene Behandlungsdauer für Patienten, die zuvor nicht mit Interferon behandelt wurden, beträgt 24 bis 48 Wochen. Die Behandlungsdauer sollte individuell auf den Patienten abgestimmt werden, abhängig von den Ausgangscharakteristika der Erkrankung, dem Ansprechen auf die Therapie und der Verträglichkeit des Regimes [siehe INDIKATIONEN UND VERWENDUNG , NEBENWIRKUNGEN , und Klinische Studien ]. Nach 24-wöchiger Behandlung sollte das virologische Ansprechen beurteilt werden. Bei allen Patienten, die innerhalb von 24 Wochen keine HCV-RNA unterhalb der Nachweisgrenze des Assays erreicht haben, sollte ein Absetzen der Behandlung in Betracht gezogen werden. Es liegen keine Sicherheits- und Wirksamkeitsdaten für eine Behandlung von mehr als 48 Wochen in der zuvor unbehandelten Patientenpopulation vor.
Dauer der Behandlung – Wiederbehandlung mit INTRON A/REBETOL bei Patienten mit Rückfall
Bei Patienten, die nach einer Monotherapie mit nichtpegyliertem Interferon einen Rückfall erleiden, beträgt die empfohlene Behandlungsdauer 24 Wochen.
Pädiatrie
Die empfohlene REBETOL-Dosis beträgt 15 mg/kg pro Tag zum Einnehmen (geteilte Dosis morgens und abends). Siehe Tabelle 2 zur pädiatrischen Dosierung von REBETOL in Kombination mit INTRON A. INTRON A zur Injektion nach Körpergewicht von 25 kg bis 61 kg beträgt 3 Millionen IE/m² dreimal wöchentlich subkutan. Siehe Dosierungstabelle für Erwachsene für mehr als 61 kg Körpergewicht.
Die empfohlene Behandlungsdauer beträgt 48 Wochen für pädiatrische Patienten mit Genotyp 1. Nach 24 Behandlungswochen sollte das virologische Ansprechen beurteilt werden. Bei allen Patienten, die bis zu diesem Zeitpunkt keine HCV-RNA unterhalb der Nachweisgrenze des Assays erreicht haben, sollte ein Absetzen der Behandlung in Erwägung gezogen werden. Die empfohlene Behandlungsdauer für pädiatrische Patienten mit Genotyp 2/3 beträgt 24 Wochen.
Labortests
Die folgenden Labortests werden für alle mit REBETOL 200 mg behandelten Patienten vor Beginn der Behandlung und danach in regelmäßigen Abständen empfohlen.
Hämatologische Standardtests – einschließlich Hämoglobin (vor der Behandlung, Woche 2 und Woche 4 der Therapie und wie klinisch angemessen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ], vollständige und differenzielle Leukozytenzahl und Thrombozytenzahl.
Dosisänderungen
Wenn während der REBETOL/INTRON A-Kombinationstherapie oder der REBETOL/PegIntron-Therapie schwerwiegende Nebenwirkungen oder Laborwertabweichungen auftreten, ändern oder setzen Sie die Dosis ab, bis die Nebenwirkung nachlässt oder an Schwere abnimmt [siehe Abschnitt 4.4]. WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Wenn die Unverträglichkeit nach einer Dosisanpassung fortbesteht, sollte die Kombinationstherapie abgebrochen werden. Die Dosisreduktion von PegIntron bei erwachsenen Patienten unter REBETOL/PegIntron-Kombinationstherapie erfolgt in einem zweistufigen Verfahren von der ursprünglichen Anfangsdosis von 1,5 µg/kg/Woche auf 1 µg/kg/Woche und dann auf 0,5 µg/kg/Woche , wenn benötigt. Weitere Informationen zur Dosisreduktion von PegIntron finden Sie auf dem Etikett von PegIntron.
In der Kombinationstherapie-Studie 2 für Erwachsene kam es bei 42 % der Patienten, die PegIntron 1,5 µg/kg und REBETOL 800 mg täglich erhielten, zu Dosisreduktionen, einschließlich 57 % der Patienten mit einem Körpergewicht von 60 kg oder weniger. In Studie 4 wurde bei 16 % der Studienteilnehmer die Dosis von PegIntron auf 1 µg/kg in Kombination mit REBETOL reduziert, wobei weitere 4 % aufgrund von Nebenwirkungen eine zweite Dosisreduktion von PegIntron auf 0,5 µg/kg benötigten [siehe NEBENWIRKUNGEN ].
Die Dosisreduktion bei pädiatrischen Patienten wird erreicht, indem die empfohlene PegIntron-Dosis in einem zweistufigen Verfahren von der ursprünglichen Anfangsdosis von 60 Mikrogramm/m²/Woche auf 40 Mikrogramm/m²/Woche und dann auf 20 Mikrogramm/m²/Woche geändert wird, falls benötigt (siehe Tabelle 4). In der pädiatrischen Studie zur Kombinationstherapie kam es bei 25 % der Patienten, die PegIntron 60 µg/m² wöchentlich und REBETOL 15 mg/kg täglich erhielten, zu Dosisreduktionen. Die Dosisreduktion bei pädiatrischen Patienten wird erreicht, indem die empfohlene REBETOL-Dosis von der ursprünglichen Anfangsdosis von 15 mg/kg täglich in einem zweistufigen Verfahren auf 12 mg/kg/Tag und dann bei Bedarf auf 8 mg/kg/Tag geändert wird ( siehe Tabelle 4).
REBETOL 200 mg sollte nicht bei Patienten mit einer Kreatinin-Clearance von weniger als 50 ml/min angewendet werden. Patienten mit eingeschränkter Nierenfunktion und solche über 50 Jahre sollten hinsichtlich der Entwicklung einer Anämie sorgfältig überwacht werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , Verwendung in bestimmten Bevölkerungsgruppen , und KLINISCHE PHARMAKOLOGIE ].
REBETOL 200 mg sollte Patienten mit vorbestehender Herzerkrankung mit Vorsicht verabreicht werden. Die Patienten sollten vor Beginn der Therapie untersucht und während der Therapie angemessen überwacht werden. Wenn es zu einer Verschlechterung des kardiovaskulären Status kommt, sollte die Therapie abgebrochen werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Bei Patienten mit stabiler kardiovaskulärer Erkrankung in der Vorgeschichte ist eine dauerhafte Dosisreduktion erforderlich, wenn der Hämoglobinwert innerhalb von 4 Wochen um mehr als oder gleich 2 g/dl abfällt. Darüber hinaus sollte bei diesen Patienten mit kardialer Vorgeschichte, wenn der Hämoglobinwert nach 4 Wochen mit reduzierter Dosis unter 12 g/dl bleibt, der Patient die Kombinationstherapie abbrechen.
Es wird empfohlen, dass bei Patienten, deren Hämoglobinspiegel unter 10 g/dl fällt, die REBETOL-Dosis gemäß Tabelle 4 angepasst oder abgesetzt wird [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Weitere Informationen zur Reduzierung einer INTRON A- oder PegIntron-Dosis finden Sie auf dem Etikett von INTRON A oder PegIntron.
Absetzen der Dosierung
Erwachsene
Bei HCV-Genotyp 1, Interferon-alfa-naiven Patienten, die PegIntron in Kombination mit Ribavirin erhalten, wird ein Absetzen der Therapie empfohlen, wenn nach 12 Wochen Therapie nicht mindestens ein Abfall oder Verlust von HCV-RNA um 2 log10 auftritt oder wenn HCV-RNA vorliegt Die Spiegel bleiben nach 24-wöchiger Therapie nachweisbar. Unabhängig vom Genotyp ist es sehr unwahrscheinlich, dass zuvor behandelte Patienten, die in Woche 12 oder 24 nachweisbare HCV-RNA aufweisen, eine SVR erreichen, und ein Absetzen der Therapie sollte in Betracht gezogen werden.
Pädiatrie (3-17 Jahre)
Es wird empfohlen, dass Patienten, die eine PegIntron/REBETOL-Kombination erhalten (außer HCV-Genotyp 2 und 3), die Therapie nach 12 Wochen absetzen, wenn ihre HCV-RNA in Behandlungswoche 12 um weniger als 2 log10 im Vergleich zu einer Vorbehandlung abgefallen ist, oder nach 24 Wochen, wenn sie nachweisbar sind HCV-RNA in Behandlungswoche 24.
WIE GELIEFERT
Darreichungsformen und Stärken
REBETOL 200 mg Kapseln 200 mg
REBETOL 200 mg Lösung zum Einnehmen 40 mg pro ml
Lagerung und Handhabung
REBETOL 200 mg Kapseln sind weiße, undurchsichtige Kapseln mit REBETOL, 200 mg, und dem Logo der Schering Corporation auf der Kapselhülle; die Kapseln sind in einer Flasche mit 56 Kapseln verpackt ( NDC 0085-1351-05), 70 Kapseln ( NDC 0085-1385-07) und 84 Kapseln ( NDC 0085-1194-03).
REBETOL 200 mg Lösung zum Einnehmen 40 mg pro ml ist eine klare, farblose bis blass- oder hellgelbe Flüssigkeit mit Kaugummigeschmack und ist in braunen 4-oz-Glasflaschen (100 ml/Flasche) mit kindersicheren Verschlüssen ( NDC 0085-1318-01).
Die Flasche mit REBETOL-Kapseln sollte bei 25 °C (77 °F) gelagert werden; Exkursionen bis 15-30°C (59-86°F) erlaubt [siehe USP kontrollierte Raumtemperatur ].
REBETOL 200 mg Lösung zum Einnehmen sollte zwischen 2-8°C (36-46°F) oder bei 25°C (77°F) gelagert werden; Exkursionen bis 15-30°C (59-86°F) erlaubt [siehe USP kontrollierte Raumtemperatur ].
REBETOL 200 mg Lösung zum Einnehmen hergestellt für: Merck Sharp & Dohme Corp., eine Tochtergesellschaft von Whitehouse Station, NJ 08889, USA. Hergestellt von: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Kanada REBETOL-Kapseln hergestellt von: Merck Sharp & Dohme Corp., eine Tochtergesellschaft von Whitehouse Station, NJ 08889, USA. Überarbeitet: Dezember 2014.
NEBENWIRKUNGEN
Klinische Studien mit REBETOL in Kombination mit PegIntron oder INTRON A wurden an über 7800 Probanden im Alter von 3 bis 76 Jahren durchgeführt.
Die primäre Toxizität von Ribavirin ist hämolytische Anämie. Reduktionen der Hämoglobinwerte traten innerhalb der ersten 1 bis 2 Wochen der oralen Therapie auf. Herz- und Lungenreaktionen im Zusammenhang mit Anämie traten bei etwa 10 % der Patienten auf [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Bei mehr als 96 % aller Probanden in klinischen Studien traten eine oder mehrere Nebenwirkungen auf. Die am häufigsten berichteten Nebenwirkungen bei erwachsenen Probanden, die PegIntron oder INTRON A in Kombination mit REBETOL 200 mg erhielten, waren Entzündungen/Reaktionen an der Injektionsstelle, Müdigkeit/Asthenie, Kopfschmerzen, Schüttelfrost, Fieber, Übelkeit, Myalgie und Angst/emotionale Labilität/Reizbarkeit. Die häufigsten Nebenwirkungen bei pädiatrischen Patienten ab 3 Jahren, die REBETOL 200 mg in Kombination mit PegIntron oder INTRON A erhielten, waren Fieber, Kopfschmerzen, Neutropenie, Müdigkeit, Anorexie, Erythem an der Injektionsstelle und Erbrechen.
Der Abschnitt „Nebenwirkungen“ bezieht sich auf die folgenden klinischen Studien:
Schwerwiegende Nebenwirkungen sind in klinischen Studien mit PegIntron mit oder ohne REBETOL bei ungefähr 12 % der Patienten aufgetreten [siehe Eingerahmte Warnung , WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die häufigsten schwerwiegenden Ereignisse, die bei Patienten auftraten, die mit PegIntron und REBETOL 200 mg behandelt wurden, waren Depressionen und Suizidgedanken [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ], die jeweils mit einer Häufigkeit von weniger als 1 % auftreten. Suizidgedanken oder -versuche traten bei pädiatrischen Patienten, vor allem Jugendlichen, häufiger auf als bei erwachsenen Patienten (2,4 % versus 1 %) während der Behandlung und Nachbeobachtung außerhalb der Therapie [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die häufigsten tödlichen Reaktionen bei Patienten, die mit PegIntron und REBETOL 200 mg behandelt wurden, waren Herzstillstand, Suizidgedanken und Suizidversuch [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ], die alle bei weniger als 1 % der Probanden auftraten.
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Raten von Nebenwirkungen nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Erfahrung aus klinischen Studien – REBETOL/PegIntron-Kombinationstherapie
Erwachsene Themen
Nebenwirkungen, die in der klinischen Studie mit einer Inzidenz von mehr als 5 % auftraten, sind in Tabelle 5 nach Behandlungsgruppe aus der REBETOL/PegIntron-Kombinationstherapie (Studie 2) aufgeführt.
Tabelle 6 fasst die behandlungsbedingten Nebenwirkungen in Studie 4 zusammen, die mit einer Inzidenz von größer oder gleich 10 % auftraten.
Die Inzidenz schwerwiegender Nebenwirkungen war in allen Studien vergleichbar. In Studie 3 wurde eine ähnliche Inzidenz schwerwiegender Nebenwirkungen für die gewichtsbasierte REBETOL-Gruppe (12 %) und für das REBETOL-Flachdosierungsschema berichtet. In Studie 2 betrug die Inzidenz schwerwiegender Nebenwirkungen 17 % in den PegIntron/REBETOL-Gruppen im Vergleich zu 14 % in der INTRON A/REBETOL-Gruppe.
In vielen, aber nicht allen Fällen verschwanden die Nebenwirkungen nach Dosisreduktion oder Absetzen der Therapie. Bei einigen Probanden traten während des 6-monatigen Nachbeobachtungszeitraums anhaltende oder neue schwerwiegende Nebenwirkungen auf. In Studie 2 traten bei vielen Probanden noch mehrere Monate nach Absetzen der Therapie Nebenwirkungen auf. Am Ende der 6-monatigen Nachbeobachtungszeit betrug die Inzidenz anhaltender Nebenwirkungen nach Körperklasse in der Gruppe mit PegIntron 1,5/REBETOL 200 mg 33 % (Psychiatrie), 20 % (Muskel-Skelett-Erkrankungen) und 10 % (für endokrine und für GI). Bei etwa 10 bis 15 % der Probanden waren Gewichtsverlust, Müdigkeit und Kopfschmerzen nicht verschwunden.
Es gab 31 Todesfälle von Probanden, die während der Behandlung oder während der Nachbeobachtung in diesen klinischen Studien auftraten. In Studie 1 gab es 1 Suizid bei einem Probanden, der PegIntron-Monotherapie erhielt, und 2 Todesfälle bei Probanden, die INTRON A-Monotherapie erhielten (1 Mord/Selbstmord und 1 plötzlicher Tod). In Studie 2 gab es 1 Suizid bei einem Probanden, der eine Kombinationstherapie mit PegIntron/REBETOL 200 mg erhielt; und 1 Probandentod in der Gruppe mit INTRON A/REBETOL 200 mg (Autounfall). In Studie 3 gab es 14 Todesfälle, von denen 2 wahrscheinlich Suizide waren und 1 ein ungeklärter Tod bei einer Person mit einer relevanten medizinischen Vorgeschichte von Depressionen war. In Studie 4 gab es 12 Todesfälle, von denen 6 bei Studienteilnehmern auftraten, die eine Kombinationstherapie mit PegIntron/REBETOL erhielten, 5 im Arm mit PegIntron 1,5 µg/REBETOL (N=1019) und 1 im Arm mit PegIntron 1 µg/REBETOL (N= 1016), von denen 6 bei Patienten auftraten, die Pegasys/Copegus erhielten (N=1035); Es gab 3 Suizide während der Nachbeobachtungszeit ohne Behandlung bei Patienten, die eine Kombinationstherapie mit PegIntron (1,5 mcg/kg)/REBETOL 200 mg erhielten.
In den Studien 1 und 2 brachen 10 bis 14 % der Patienten, die PegIntron allein oder in Kombination mit REBETOL 200 mg erhielten, die Therapie ab, verglichen mit 6 %, die mit INTRON A allein behandelt wurden, und 13 %, die mit INTRON A in Kombination mit REBETOL behandelt wurden. In ähnlicher Weise brachen in Studie 3 15 % der Patienten, die PegIntron in Kombination mit gewichtsbasiertem REBETOL erhielten, und 14 % der Patienten, die PegIntron und 200 mg REBETOL in fester Dosis erhielten, die Therapie aufgrund einer Nebenwirkung ab. Die häufigsten Gründe für das Absetzen der Therapie standen im Zusammenhang mit bekannten Interferon-Wirkungen von psychiatrischen, systemischen (z. B. Müdigkeit, Kopfschmerzen) oder gastrointestinalen Nebenwirkungen. In Studie 4 brachen 13 % der Patienten im Arm mit PegIntron 1,5 µg/REBETOL 200 mg, 10 % im Arm mit PegIntron 1 µg/REBETOL 200 mg und 13 % im Arm mit Pegasys 180 µg/Copegus die Behandlung aufgrund von Nebenwirkungen ab.
In Studie 2 kam es bei 42 % der Patienten, die PegIntron (1,5 µg/kg)/REBETOL 200 mg erhielten, und bei 34 % der Patienten, die INTRON A/REBETOL erhielten, zu Dosisreduktionen aufgrund von Nebenwirkungen. Die Mehrheit der Probanden (57 %) mit einem Gewicht von 60 kg oder weniger, die PegIntron (1,5 mcg/kg)/REBETOL erhielten, erforderte eine Dosisreduktion. Die Reduktion von Interferon war dosisabhängig (PegIntron 1,5 µg/kg größer als PegIntron 0,5 µg/kg oder INTRON A), 40 %, 27 % bzw. 28 %. Die Dosisreduktion für REBETOL 200 mg war in allen drei Gruppen ähnlich, 33 bis 35 %. Die häufigsten Gründe für Dosisanpassungen waren Neutropenie (18 %) oder Anämie (9 %) (vgl Laborwerte ). Andere häufige Gründe waren Depressionen, Müdigkeit, Übelkeit und Thrombozytopenie. In Studie 3 traten Dosisanpassungen aufgrund von Nebenwirkungen bei gewichtsbasierter Dosierung (WBD) häufiger auf als bei unveränderter Dosierung (29 % bzw. 23 %). In Studie 4 erhielten 16 % der Studienteilnehmer eine Dosisreduktion von PegIntron auf 1 µg/kg in Kombination mit REBETOL 200 mg, wobei weitere 4 % aufgrund von Nebenwirkungen die zweite Dosisreduktion von PegIntron auf 0,5 µg/kg benötigten, verglichen mit 15 % der Patienten im Pegasys/Copegus-Arm, die eine Dosisreduktion auf 135 µg/Woche mit Pegasys benötigten, wobei weitere 7 % im Pegasys/Copegus-Arm eine zweite Dosisreduktion auf 90 µg/Woche mit Pegasys benötigten.
In den Kombinationsstudien mit PegIntron/REBETOL 200 mg waren die häufigsten Nebenwirkungen psychiatrischer Natur, die bei 77 % der Studienteilnehmer in Studie 2 und bei 68 % bis 69 % der Studienteilnehmer in Studie 3 auftraten. Diese psychiatrischen Nebenwirkungen waren am häufigsten Depression, Reizbarkeit und Schlaflosigkeit, die jeweils von etwa 30 % bis 40 % der Patienten in allen Behandlungsgruppen berichtet wurden. Suizidverhalten (Ideen, Versuche und Suizide) trat bei 2 % aller Probanden während der Behandlung oder während der Nachbeobachtung nach Beendigung der Behandlung auf [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. In Studie 4 traten psychiatrische Nebenwirkungen bei 58 % der Studienteilnehmer im PegIntron 1,5 µg/REBETOL 200 mg-Arm, bei 55 % der Studienteilnehmer im PegIntron 1 µg/REBETOL 200 mg-Arm und bei 57 % der Studienteilnehmer im Pegasys 180 µg/Copegus-Arm auf .
PegIntron verursachte bei etwa zwei Dritteln der Probanden Müdigkeit oder Kopfschmerzen, bei etwa der Hälfte der Probanden Fieber oder Schüttelfrost. Die Schwere einiger dieser systemischen Symptome (z. B. Fieber und Kopfschmerzen) nahm im Laufe der Behandlung tendenziell ab. In den Studien 1 und 2 traten Entzündungen und Reaktionen an der Applikationsstelle (z. B. Blutergüsse, Juckreiz und Reizungen) bei PegIntron-Therapien etwa doppelt so häufig auf (bei bis zu 75 % der Patienten) im Vergleich zu INTRON A. Schmerzen an der Injektionsstelle traten jedoch auf selten (2 bis 3 %) in allen Gruppen. In Studie 3 gab es insgesamt eine Inzidenz von 23 % bis 24 % für Reaktionen oder Entzündungen an der Injektionsstelle.
Patienten, die REBETOL/PegIntron als Wiederbehandlung nach Versagen einer vorherigen Interferon-Kombinationsbehandlung erhielten, berichteten in klinischen Studien mit nicht vorbehandelten Patienten über Nebenwirkungen, die denen ähnelten, die zuvor mit dieser Behandlung in Verbindung gebracht wurden.
Pädiatrische Fächer
Im Allgemeinen war das Nebenwirkungsprofil bei Kindern und Jugendlichen ähnlich wie bei Erwachsenen. In der pädiatrischen Studie waren die häufigsten Nebenwirkungen bei allen Probanden Fieber (80 %), Kopfschmerzen (62 %), Neutropenie (33 %), Müdigkeit (30 %), Anorexie (29 %), Erythem an der Injektionsstelle (29 %) und Erbrechen (27 %). Die Mehrheit der in der Studie berichteten Nebenwirkungen war leicht oder mittelschwer. Schwere Nebenwirkungen wurden bei 7 % (8/107) aller Probanden berichtet und umfassten Schmerzen an der Injektionsstelle (1 %), Gliederschmerzen (1 %), Kopfschmerzen (1 %), Neutropenie (1 %) und Fieber (4 %). Wichtige Nebenwirkungen, die bei dieser Patientenpopulation auftraten, waren Nervosität (7 %; 7/107), Aggression (3 %; 3/107), Wut (2 %; 2/107) und Depression (1 %; 1/107). . Fünf Probanden erhielten eine Behandlung mit Levothyroxin, drei mit klinischer Hypothyreose und zwei mit asymptomatischen TSH-Erhöhungen. Die Gewichts- und Größenzunahme pädiatrischer Probanden, die mit PegIntron plus REBETOL behandelt wurden, blieb über die gesamte Behandlungsdauer hinter den Prognosen der normativen Populationsdaten zurück. Während der Behandlung wurde bei 70 % der Probanden eine stark gehemmte Wachstumsgeschwindigkeit (weniger als 3. Perzentil) beobachtet.
Dosisanpassungen von PegIntron und/oder Ribavirin waren bei 25 % der Patienten aufgrund von behandlungsbedingten Nebenwirkungen erforderlich, am häufigsten bei Anämie, Neutropenie und Gewichtsverlust. Zwei Probanden (2 %; 2/107) brachen die Therapie aufgrund einer Nebenwirkung ab.
Nebenwirkungen, die mit einer Inzidenz von größer oder gleich 10 % bei den pädiatrischen Studienteilnehmern auftraten, sind in Tabelle 7 aufgeführt.
94 von 107 Probanden nahmen an einer 5-jährigen Langzeit-Follow-up-Studie teil. Die langfristigen Wirkungen auf das Wachstum waren bei den 24 Wochen behandelten Probanden geringer als bei den 48 Wochen behandelten. 24 % der Patienten (11/46), die 24 Wochen lang behandelt wurden, und 40 % der Patienten (19/48), die 48 Wochen lang behandelt wurden, hatten eine Altersabnahme von > 15 Perzentilen von der Vorbehandlung bis zum Ende des 5. Jahres Langzeit-Follow-up im Vergleich zu Baseline-Perzentilen vor der Behandlung. Bei elf Prozent der Patienten (5/46), die 24 Wochen behandelt wurden, und bei 13 % der Patienten (6/48), die 48 Wochen behandelt wurden, wurde eine Abnahme gegenüber dem Ausgangswert vor der Behandlung von > 30 altersbezogenen Perzentilen der Körpergröße bis zum Ende beobachtet der 5-Jahres-Langzeit-Follow-up. Obwohl in allen Altersgruppen beobachtet, schien das höchste Risiko für eine reduzierte Körpergröße am Ende der Langzeitnachbeobachtung mit dem Beginn der Kombinationstherapie in den Jahren der erwarteten maximalen Wachstumsgeschwindigkeit zu korrelieren. [Sehen WARNUNGEN UND VORSICHTSMASSNAHMEN ]
Laborwerte
Erwachsene und pädiatrische Themen
Das Nebenwirkungsprofil in Studie 3, in der die Kombination aus PegIntron/gewichtsbasiertem REBETOL 200 mg mit einer Kombination aus PegIntron/ REBETOL 200 mg als Einheitsdosis verglichen wurde, zeigte eine erhöhte Anämierate bei gewichtsbasierter Dosierung (29 % vs. 19 % bei gewichtsbasierter Dosierung). vs. Flat-Dosis-Schemata). Die Mehrzahl der Fälle von Anämie verlief jedoch leicht und sprach auf Dosisreduktionen an.
Nachfolgend werden die Veränderungen ausgewählter Laborwerte während der Behandlung in Kombination mit REBETOL 200 mg beschrieben. Abnahmen von Hämoglobin, Leukozyten, Neutrophilen und Blutplättchen können eine Dosisreduktion oder ein dauerhaftes Absetzen der Therapie erforderlich machen [sehen DOSIERUNG UND ANWENDUNG ]. Die Veränderungen ausgewählter Laborwerte während der Therapie sind in Tabelle 8 beschrieben. Die meisten Veränderungen der Laborwerte in der PegIntron/REBETOL 200 mg-Studie mit Kindern und Jugendlichen waren leicht oder mittelschwer.
Hämoglobin
Der Hämoglobinspiegel sank bei etwa 30 % der Probanden in Studie 2 auf weniger als 11 g/dl. In Studie 3 wiesen 47 % der Probanden, die WBD REBETOL 200 mg erhielten, und 33 % der Probanden, die REBETOL 200 mg als Flachdosis erhielten, einen Abfall des Hämoglobinspiegels um weniger als 11 g auf /dl. Senkungen des Hämoglobinwertes auf weniger als 9 g/dl traten häufiger bei Patienten auf, die WBD erhielten, verglichen mit einer flachen Dosierung (4 % bzw. 2 %). In Studie 2 war bei 9 % bzw. 13 % der Patienten in den Gruppen mit PegIntron/REBETOL 200 mg und INTRON A/REBETOL 200 mg eine Dosisanpassung erforderlich. In Studie 4 kam es bei Probanden, die PegIntron (1,5 mcg/kg)/REBETOL erhielten, zu einem Abfall der Hämoglobinwerte auf 8,5 bis weniger als 10 g/dl (28 %) und auf weniger als 8,5 g/dl (3 %), wohingegen bei Patienten Bei Erhalt von Pegasys 180 mcg/Copegus traten diese Abnahmen bei 26 % bzw. 4 % der Studienteilnehmer auf. Die Hämoglobinspiegel wurden im Durchschnitt in den Behandlungswochen 4–6 stabil. Das beobachtete typische Muster war eine Abnahme der Hämoglobinspiegel bis Behandlungswoche 4, gefolgt von einer Stabilisierung und einem Plateau, das bis zum Ende der Behandlung aufrechterhalten wurde. In der PegIntron-Monotherapiestudie war der Hämoglobinabfall im Allgemeinen leicht und Dosisanpassungen waren selten erforderlich [siehe DOSIERUNG UND ANWENDUNG ].
Neutrophile
Bei der Mehrzahl der erwachsenen Patienten, die in Studie 2 eine Kombinationstherapie mit 200 mg REBETOL und INTRON A/REBETOL (60 %) erhielten, wurde eine Abnahme der Neutrophilenzahl beobachtet. Eine schwere potenziell lebensbedrohliche Neutropenie (weniger als 0,5 x 109/l) trat bei 2 % der mit INTRON A/REBETOL 200 mg behandelten Patienten und bei etwa 4 % der mit PegIntron/REBETOL 200 mg behandelten Patienten in Studie 2 auf. Achtzehn Prozent der Patienten erhielten PegIntron/REBETOL in Studie 2 erforderte eine Änderung der Interferon-Dosierung. Nur wenige Patienten (weniger als 1 %) mussten die Behandlung dauerhaft abbrechen. Die Neutrophilenzahl kehrte im Allgemeinen 4 Wochen nach Beendigung der Therapie auf das Niveau vor der Behandlung zurück [siehe DOSIERUNG UND ANWENDUNG ].
Blutplättchen
Bei etwa 20 % der mit PegIntron allein oder mit 200 mg REBETOL behandelten Patienten und bei 6 % der mit INTRON A/REBETOL behandelten erwachsenen Patienten sank die Thrombozytenzahl auf weniger als 100.000/mm³. Ein starker Rückgang der Thrombozytenzahl (weniger als 50.000/mm³) tritt bei weniger als 4 % der erwachsenen Probanden auf. Patienten können aufgrund einer Abnahme der Thrombozytenzahl ein Absetzen oder eine Dosisanpassung benötigen [siehe DOSIERUNG UND ANWENDUNG ]. In Studie 2 benötigten 1 % bzw. 3 % der Studienteilnehmer eine Dosisanpassung von INTRON A bzw. PegIntron. Die Thrombozytenzahl kehrte im Allgemeinen 4 Wochen nach Beendigung der Therapie auf das Niveau vor der Behandlung zurück.
Schilddrüsenfunktion
Die Entwicklung von TSH-Anomalien, mit oder ohne klinische Manifestationen, ist mit Interferontherapien verbunden. In Studie 2 traten klinisch offensichtliche Schilddrüsenerkrankungen bei Patienten auf, die entweder mit INTRON A oder PegIntron (mit oder ohne REBETOL) behandelt wurden, mit einer ähnlichen Häufigkeit (5 % für Hypothyreose und 3 % für Hyperthyreose). Die Probanden entwickelten neu auftretende TSH-Anomalien während der Behandlung und während des Nachbeobachtungszeitraums. Am Ende des Nachbeobachtungszeitraums hatten noch 7 % der Probanden abnorme TSH-Werte.
Bilirubin und Harnsäure
In Studie 2 entwickelten 10 bis 14 % der Probanden eine Hyperbilirubinämie und 33 bis 38 % eine Hyperurikämie in Verbindung mit Hämolyse. Sechs Probanden entwickelten eine leichte bis mittelschwere Gicht.
Erfahrung aus klinischen Studien – REBETOL/INTRON Eine Kombinationstherapie
Erwachsene Themen
In klinischen Studien brachen 19 % bzw. 6 % der zuvor unbehandelten bzw. Patienten mit Rückfall die Therapie aufgrund von Nebenwirkungen in den Kombinationsarmen ab, verglichen mit 13 % bzw. 3 % in den Interferon-Armen. Ausgewählte behandlungsbedingte Nebenwirkungen, die in den US-Studien mit einer Inzidenz von mindestens 5 % auftraten, sind nach Behandlungsgruppe aufgeführt (siehe Tabelle 9). Im Allgemeinen wurden die ausgewählten behandlungsbedingten Nebenwirkungen in den internationalen Studien im Vergleich zu den US-Studien mit geringerer Inzidenz berichtet, mit Ausnahme von Asthenie, grippeähnlichen Symptomen, Nervosität und Juckreiz.
Pädiatrische Fächer
In klinischen Studien mit 118 pädiatrischen Probanden im Alter von 3 bis 16 Jahren brachen 6 % die Therapie aufgrund von Nebenwirkungen ab. Dosisanpassungen waren bei 30 % der Studienteilnehmer erforderlich, am häufigsten bei Anämie und Neutropenie. Im Allgemeinen war das Nebenwirkungsprofil bei Kindern und Jugendlichen ähnlich wie bei Erwachsenen. Störungen an der Injektionsstelle, Fieber, Anorexie, Erbrechen und emotionale Labilität traten bei pädiatrischen Probanden häufiger auf als bei erwachsenen Probanden. Umgekehrt erlebten pädiatrische Probanden im Vergleich zu erwachsenen Probanden weniger Müdigkeit, Dyspepsie, Arthralgie, Schlaflosigkeit, Reizbarkeit, Konzentrationsstörungen, Dyspnoe und Juckreiz. Ausgewählte behandlungsbedingte Nebenwirkungen, die bei allen pädiatrischen Patienten, die die empfohlene Dosis der REBETOL/INTRON A-Kombinationstherapie erhielten, mit einer Häufigkeit von mindestens 5 % auftraten, sind in Tabelle 9 aufgeführt.
Während eines 48-wöchigen Therapieverlaufs kam es zu einer Abnahme der Rate des linearen Wachstums (Abnahme der mittleren Perzentilzuordnung von 7 %) und einer Abnahme der Gewichtszunahme (Abnahme der mittleren Perzentilzuordnung von 9 %). Während der 24-wöchigen Nachbehandlungszeit wurde eine allgemeine Umkehrung dieser Trends festgestellt. Langzeitdaten bei einer begrenzten Anzahl von Patienten deuten jedoch darauf hin, dass die Kombinationstherapie eine Wachstumshemmung hervorrufen kann, die bei einigen Patienten zu einer verringerten Endgröße im Erwachsenenalter führt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Laborwerte
Änderungen ausgewählter hämatologischer Werte (Hämoglobin, weiße Blutkörperchen, Neutrophile und Blutplättchen) während der Therapie sind unten beschrieben (siehe Tabelle 10).
Hämoglobin. Hämoglobinabnahmen bei Patienten, die eine REBETOL-Therapie erhielten, begannen in Woche 1, mit einer Stabilisierung bis Woche 4. Bei zuvor unbehandelten Patienten, die 48 Wochen lang behandelt wurden, betrug die mittlere maximale Abnahme gegenüber dem Ausgangswert 3,1 g/dl in der US-Studie und 2,9 g/dl in der internationalen Studie Studie. Bei Patienten mit Rückfall betrug die mittlere maximale Abnahme gegenüber dem Ausgangswert 2,8 g/dl in der US-Studie und 2,6 g/dl in der internationalen Studie. Die Hämoglobinwerte kehrten bei den meisten Probanden innerhalb von 4 bis 8 Wochen nach Beendigung der Therapie auf die Werte vor der Behandlung zurück.
Bilirubin und Harnsäure. In klinischen Studien wurden Anstiege von Bilirubin und Harnsäure in Verbindung mit Hämolyse festgestellt. Bei den meisten handelte es sich um moderate biochemische Veränderungen, die innerhalb von 4 Wochen nach Absetzen der Behandlung rückgängig gemacht wurden. Diese Beobachtung trat am häufigsten bei Patienten mit einer früheren Diagnose des Gilbert-Syndroms auf. Dies wurde nicht mit Leberfunktionsstörungen oder klinischer Morbidität in Verbindung gebracht.
Postmarketing-Erfahrungen
Die folgenden Nebenwirkungen wurden während der Anwendung von REBETOL in Kombination mit INTRON A oder PegIntron nach der Zulassung festgestellt und berichtet. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Erkrankungen des Blutes und des Lymphsystems
Aplasie der reinen roten Blutkörperchen, aplastische Anämie
Erkrankungen des Ohrs und des Labyrinths
Hörstörung, Schwindel
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
Pulmonale Hypertonie
Augenerkrankungen
Seröse Netzhautablösung
Endokrine Störungen
Diabetes
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Didanosin
Die Exposition gegenüber Didanosin oder seinem aktiven Metaboliten (Didesoxyadenosin-5'-triphosphat) ist erhöht, wenn Didanosin zusammen mit Ribavirin verabreicht wird, was klinische Toxizitäten verursachen oder verschlimmern könnte; daher ist die gleichzeitige Anwendung von REBETOL 200 mg Kapseln oder Lösung zum Einnehmen und Didanosin kontraindiziert. In klinischen Studien wurde über Fälle von tödlichem Leberversagen sowie peripherer Neuropathie, Pankreatitis und symptomatischer Hyperlaktatämie/Laktazidose berichtet.
Nucleosid-Analoga
Leberdekompensation (einige mit tödlichem Ausgang) ist bei zirrhotischen HIV/HCV-koinfizierten Patienten aufgetreten, die eine antiretrovirale Kombinationstherapie gegen HIV und Interferon alpha und Ribavirin erhielten. Eine zusätzliche Behandlung mit alpha-Interferonen allein oder in Kombination mit Ribavirin kann das Risiko in dieser Patientengruppe erhöhen. Patienten, die Interferon mit Ribavirin und Nukleosid-Reverse-Transkriptase-Hemmern (NRTIs) erhalten, sollten engmaschig auf behandlungsassoziierte Toxizitäten, insbesondere Leberdekompensation und Anämie, überwacht werden. Das Absetzen von NRTIs sollte als medizinisch angemessen betrachtet werden (siehe Kennzeichnung für einzelne NRTI-Produkte ). Eine Dosisreduktion oder das Absetzen von Interferon, Ribavirin oder beiden sollte ebenfalls in Betracht gezogen werden, wenn eine Verschlechterung der klinischen Toxizität beobachtet wird, einschließlich Leberdekompensation (z. B. Child-Pugh größer als 6).
Ribavirin kann die antivirale Aktivität von Stavudin und Zidovudin in Zellkulturen gegen HIV antagonisieren. In Zellkulturen wurde gezeigt, dass Ribavirin die Phosphorylierung von Lamivudin, Stavudin und Zidovudin hemmt, was zu einer verringerten antiretroviralen Aktivität führen könnte. In einer Studie mit einem anderen pegylierten Interferon in Kombination mit Ribavirin wurden jedoch keine pharmakokinetischen (z. B. Plasmakonzentrationen oder intrazelluläre Konzentrationen von triphosphorylierten aktiven Metaboliten) oder pharmakodynamischen (z. B. Verlust der virologischen Suppression von HIV/HCV) Wechselwirkungen beobachtet, wenn Ribavirin und Lamivudin (n =18), Stavudin (n=10) oder Zidovudin (n=6) wurden als Teil einer Multidrug-Therapie bei HIV/HCV-koinfizierten Probanden gleichzeitig verabreicht. Daher sollte die gleichzeitige Anwendung von Ribavirin mit einem dieser Arzneimittel mit Vorsicht erfolgen.
Medikamente, die durch Cytochrom P-450 metabolisiert werden
Die Ergebnisse von In-vitro-Studien unter Verwendung von Mikrosomenpräparaten aus menschlicher und Rattenleber zeigten einen geringen oder keinen Cytochrom-P-450-Enzym-vermittelten Metabolismus von Ribavirin mit einem minimalen Potenzial für P-450-Enzym-basierte Arzneimittelwechselwirkungen.
In einer pharmakokinetischen Studie mit Mehrfachgabe wurden keine pharmakokinetischen Wechselwirkungen zwischen INTRON A und REBETOL 200 mg Kapseln festgestellt.
Azathioprin
Es wurde berichtet, dass die Anwendung von Ribavirin zur Behandlung von chronischer Hepatitis C bei Patienten, die Azathioprin erhalten, eine schwere Panzytopenie hervorruft und das Risiko einer Azathioprin-bedingten Myelotoxizität erhöhen kann. Inosinmonophosphatdehydrogenase (IMDH) wird für einen der Stoffwechselwege von Azathioprin benötigt. Es ist bekannt, dass Ribavirin IMDH hemmt, was zur Akkumulation eines Azathioprin-Metaboliten, 6-Methylthioinosinmonophosphat (6-MTITP), führt, der mit Myelotoxizität (Neutropenie, Thrombozytopenie und Anämie) assoziiert ist. Bei Patienten, die Azathioprin mit Ribavirin erhalten, sollte ein vollständiges Blutbild, einschließlich Thrombozytenzahl, im ersten Monat wöchentlich, im zweiten und dritten Behandlungsmonat zweimal monatlich, dann monatlich oder häufiger, wenn Dosis- oder andere Therapieänderungen erforderlich sind, kontrolliert werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Schwangerschaft
REBETOL 200 mg Kapseln und Lösung zum Einnehmen können Geburtsfehler und den Tod des ungeborenen Kindes verursachen. Mit der REBETOL-Therapie sollte erst begonnen werden, wenn unmittelbar vor dem geplanten Therapiebeginn ein Bericht über einen negativen Schwangerschaftstest vorliegt. Patientinnen sollten während der Behandlung und während der 6 Monate nach Beendigung der Behandlung mindestens zwei Formen der Empfängnisverhütung anwenden und monatliche Schwangerschaftstests durchführen lassen. Es muss äußerste Sorgfalt darauf verwendet werden, eine Schwangerschaft bei weiblichen Patienten und bei weiblichen Partnern von männlichen Patienten zu vermeiden. REBETOL 200mg hat gezeigt signifikante teratogene und embryozide Wirkungen bei allen Tierarten, bei denen entsprechende Studien durchgeführt wurden. Diese Wirkungen traten bei Dosen wie auf nur ein Zwanzigstel der für den Menschen empfohlenen Ribavirin-Dosis. Die Behandlung mit REBETOL 200 mg sollte erst begonnen werden, wenn ein negativer Schwangerschaftstest vorliegt unmittelbar vor dem geplanten Therapiebeginn erhalten wurde [vgl Eingerahmte Warnung , KONTRAINDIKATIONEN , Verwendung in bestimmten Bevölkerungsgruppen , und INFORMATIONEN ZUM PATIENTEN ].
Anämie
Die primäre Toxizität von Ribavirin ist hämolytische Anämie, die in klinischen Studien bei etwa 10 % der mit REBETOL/INTRON A behandelten Patienten beobachtet wurde. Die mit REBETOL-Kapseln verbundene Anämie tritt innerhalb von 1 bis 2 Wochen nach Beginn der Therapie auf. Da der anfängliche Hämoglobinabfall signifikant sein kann, wird empfohlen, Hämoglobin oder Hämatokrit vor Beginn der Behandlung und in Woche 2 und Woche 4 der Therapie oder häufiger, wenn klinisch angezeigt, zu bestimmen. Die Patienten sollten dann wie klinisch angemessen überwacht werden [siehe DOSIERUNG UND ANWENDUNG ].
Bei Patienten mit durch REBETOL verursachter Anämie wurde über tödliche und nicht tödliche Myokardinfarkte berichtet. Die Patienten sollten vor Beginn der Ribavirin-Therapie auf zugrunde liegende Herzerkrankungen untersucht werden. Bei Patienten mit vorbestehender Herzerkrankung sollten vor der Behandlung Elektrokardiogramme erstellt und während der Therapie angemessen überwacht werden. Wenn sich der kardiovaskuläre Zustand verschlechtert, sollte die Therapie ausgesetzt oder abgebrochen werden [siehe DOSIERUNG UND ANWENDUNG ]. Da eine Herzerkrankung durch eine arzneimittelinduzierte Anämie verschlimmert werden kann, sollten Patienten mit einer signifikanten oder instabilen Herzerkrankung in der Vorgeschichte REBETOL nicht anwenden.
Pankreatitis
Die Therapie mit REBETOL und INTRON A oder PegIntron sollte bei Patienten mit Anzeichen und Symptomen einer Pankreatitis ausgesetzt und bei Patienten mit bestätigter Pankreatitis abgebrochen werden.
Lungenerkrankungen
Lungensymptome, einschließlich Dyspnoe, Lungeninfiltrate, Pneumonitis, pulmonale Hypertonie und Pneumonie, wurden während der Therapie mit REBETOL in Kombination mit Alpha-Interferon berichtet; gelegentliche Fälle von tödlicher Lungenentzündung sind aufgetreten. Darüber hinaus wurde über Sarkoidose oder die Exazerbation einer Sarkoidose berichtet. Bei Anzeichen von Lungeninfiltraten oder Lungenfunktionsstörungen sollte der Patient engmaschig überwacht und gegebenenfalls die Kombinationstherapie abgebrochen werden.
Augenerkrankungen
Ribavirin wird in Kombinationstherapie mit Alpha-Interferonen angewendet. Abnahme oder Verlust des Sehvermögens, Retinopathie einschließlich Makulaödem, Netzhautarterie oder -vene, Thrombose, Netzhautblutungen und Cotton-Wool-Flecken, Optikusneuritis, Papillenödem und seröse Netzhautablösung werden durch die Behandlung mit alpha-Interferonen induziert oder verschlimmert. Alle Patienten sollten zu Studienbeginn eine Augenuntersuchung erhalten. Patienten mit vorbestehenden Augenerkrankungen (z. B. diabetische oder hypertensive Retinopathie) sollten während der Kombinationstherapie mit Alpha-Interferon regelmäßig augenärztlich untersucht werden. Jeder Patient, der Augensymptome entwickelt, sollte umgehend einer vollständigen Augenuntersuchung unterzogen werden. Die Kombinationstherapie mit alpha-Interferonen sollte bei Patienten, die neue oder sich verschlechternde ophthalmologische Erkrankungen entwickeln, abgesetzt werden.
Labortests
PegIntron in Kombination mit Ribavirin kann eine starke Abnahme der Neutrophilen- und Thrombozytenzahl sowie hämatologische, endokrine (z. B. TSH) und hepatische Anomalien verursachen.
Bei Patienten unter PegIntron/REBETOL-Kombinationstherapie sollten vor Beginn der Behandlung und danach in regelmäßigen Abständen hämatologische und blutchemische Untersuchungen durchgeführt werden. In der klinischen Studie mit Erwachsenen wurden während des Behandlungszeitraums in den Wochen 2, 4, 8, 12 und 20 das komplette Blutbild (einschließlich Hämoglobin, Neutrophilen und Thrombozytenzahl) und Blutwerte (einschließlich AST, ALT, Bilirubin und Harnsäure) gemessen dann in 6-wöchigen Abständen oder häufiger, wenn Anomalien aufgetreten sind. Bei pädiatrischen Probanden wurden die gleichen Laborparameter mit zusätzlicher Bestimmung des Hämoglobins in Behandlungswoche 6 ausgewertet. Die TSH-Werte wurden alle 12 Wochen während der Behandlungsdauer gemessen. HCV-RNA sollte während der Behandlung regelmäßig gemessen werden [siehe DOSIERUNG UND ANWENDUNG ].
Zahn- und Parodontalerkrankungen
Bei Patienten, die eine Kombinationstherapie aus Ribavirin und Interferon oder Peginterferon erhielten, wurden Zahn- und Parodontalerkrankungen berichtet. Darüber hinaus könnte Mundtrockenheit während einer Langzeitbehandlung mit der Kombination von REBETOL und pegyliertem oder nicht pegyliertem Interferon alfa-2b eine schädigende Wirkung auf Zähne und Mundschleimhäute haben. Die Patienten sollten ihre Zähne zweimal täglich gründlich putzen und sich regelmäßig zahnärztlich untersuchen lassen. Wenn Erbrechen auftritt, sollten sie darauf hingewiesen werden, den Mund danach gründlich auszuspülen.
Gleichzeitige Verabreichung von Azathioprin
In der Literatur wurde über das Auftreten von Panzytopenie (deutliche Abnahme der roten Blutkörperchen, Neutrophilen und Blutplättchen) und Knochenmarksuppression innerhalb von 3 bis 7 Wochen nach gleichzeitiger Verabreichung von pegyliertem Interferon/Ribavirin und Azathioprin berichtet. Bei dieser begrenzten Anzahl von Patienten (n = 8) war die Myelotoxizität innerhalb von 4 bis 6 Wochen nach Absetzen sowohl der antiviralen HCV-Therapie als auch der begleitenden Azathioprin-Therapie reversibel und trat bei Wiederaufnahme einer der beiden Behandlungen allein nicht wieder auf. PegIntron, REBETOL und Azathioprin sollten bei Panzytopenie abgesetzt werden, und pegyliertes Interferon/Ribavirin sollte nicht zusammen mit Azathioprin wieder eingeführt werden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Auswirkungen auf das Wachstum – Verwendung in der Pädiatrie
Die Daten zu den Wirkungen von PegIntron und REBETOL auf das Wachstum stammen aus einer offenen Studie an Probanden im Alter von 3 bis 17 Jahren, in der Gewichts- und Größenänderungen mit normativen US-Bevölkerungsdaten verglichen wurden. Im Allgemeinen bleibt die Gewichts- und Größenzunahme von mit PegIntron und REBETOL 200 mg behandelten pädiatrischen Probanden über die gesamte Behandlungsdauer hinter den Vorhersagen der normativen Populationsdaten zurück. Während der Behandlung wurde bei 70 % der Probanden eine stark gehemmte Wachstumsgeschwindigkeit (weniger als 3. Perzentil) beobachtet. Nach der Behandlung traten bei den meisten Probanden Rebound-Wachstum und Gewichtszunahme auf. Langzeit-Follow-up-Daten bei pädiatrischen Probanden weisen jedoch darauf hin, dass PegIntron in Kombinationstherapie mit REBETOL eine Wachstumshemmung hervorrufen kann, die bei einigen Patienten zu einer verringerten Körpergröße im Erwachsenenalter führt [siehe NEBENWIRKUNGEN ].
In ähnlicher Weise wurde eine Auswirkung auf das Wachstum bei Probanden nach einer einjährigen Behandlung mit REBETOL 200 mg und einer INTRON A-Kombinationstherapie beobachtet. In einer Langzeit-Follow-up-Studie mit einer begrenzten Anzahl dieser Probanden führte die Kombinationstherapie bei einigen Probanden zu einer reduzierten endgültigen Erwachsenengröße [siehe NEBENWIRKUNGEN ].
Nutzungsschutz
Basierend auf den Ergebnissen klinischer Studien ist eine Ribavirin-Monotherapie zur Behandlung einer chronischen Hepatitis-C-Virusinfektion nicht wirksam; Daher dürfen REBETOL 200 mg Kapseln oder Lösung zum Einnehmen nicht allein verwendet werden. Die Sicherheit und Wirksamkeit von REBETOL Kapseln und Lösung zum Einnehmen wurde nur bei Anwendung zusammen mit INTRON A oder PegIntron (nicht anderen Interferonen) als Kombinationstherapie nachgewiesen.
Die Sicherheit und Wirksamkeit der Therapie mit REBETOL/INTRON A und PegIntron zur Behandlung von HIV-Infektionen, Adenovirus-, RSV-, Parainfluenza- oder Influenza-Infektionen wurde nicht nachgewiesen. REBETOL 200 mg Kapseln sollten bei diesen Indikationen nicht angewendet werden. Ribavirin zur Inhalation hat eine separate Kennzeichnung, die zu Rate gezogen werden sollte, wenn eine Inhalationstherapie mit Ribavirin in Betracht gezogen wird.
Es gibt signifikante Nebenwirkungen, die durch die Therapie mit REBETOL/INTRON A oder PegIntron verursacht werden, einschließlich schwerer Depression und Suizidgedanken, hämolytische Anämie, Unterdrückung der Knochenmarkfunktion, Autoimmun- und Infektionskrankheiten, Lungenfunktionsstörung, Pankreatitis und Diabetes. Suizidgedanken oder -versuche traten bei pädiatrischen Patienten, hauptsächlich Jugendlichen, häufiger auf als bei erwachsenen Patienten (2,4 % gegenüber 1 %) während der Behandlung und während der Nachbeobachtung außerhalb der Therapie. Die Kennzeichnung von INTRON A und PegIntron sollte vor Beginn einer Kombinationsbehandlung vollständig auf zusätzliche Sicherheitsinformationen überprüft werden.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( Leitfaden für Medikamente ).
Anämie
Die häufigste Nebenwirkung von REBETOL-Kapseln ist Anämie, die schwerwiegend sein kann [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ]. Die Patienten sollten darauf hingewiesen werden, dass Laboruntersuchungen vor Beginn der Therapie und danach in regelmäßigen Abständen erforderlich sind [siehe DOSIERUNG UND ANWENDUNG ]. Es wird empfohlen, dass die Patienten gut hydriert sind, insbesondere während der Anfangsphase der Behandlung.
Schwangerschaft
Die Patienten müssen darüber informiert werden, dass REBETOL 200 mg Kapseln und Lösung zum Einnehmen Geburtsfehler und den Tod des ungeborenen Kindes verursachen können. REBETOL 200 mg darf nicht von schwangeren Frauen oder von Männern mit schwangeren Partnerinnen eingenommen werden. Bei Patientinnen und Partnerinnen von männlichen Patienten, die REBETOL einnehmen, ist äußerste Vorsicht geboten, um eine Schwangerschaft zu vermeiden. Mit REBETOL sollte erst begonnen werden, wenn unmittelbar vor Therapiebeginn ein Bericht über einen negativen Schwangerschaftstest vorliegt. Die Patientinnen müssen während der Therapie und für 6 Monate nach der Therapie monatlich einen Schwangerschaftstest durchführen.
Frauen im gebärfähigen Alter müssen vor Beginn der Therapie über die Anwendung einer wirksamen Empfängnisverhütung (zwei zuverlässige Formen) aufgeklärt werden. Patienten (männlich und weiblich) müssen auf die teratogenen/embryoziden Risiken hingewiesen und angewiesen werden, während REBETOL 200 mg und für 6 Monate nach der Therapie eine wirksame Empfängnisverhütung anzuwenden. Patienten (männlich und weiblich) sollten angewiesen werden, im Falle einer Schwangerschaft unverzüglich den Arzt zu benachrichtigen [siehe KONTRAINDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , und Verwendung in bestimmten Populationen ].
Wenn während der Behandlung oder innerhalb von 6 Monaten nach der Therapie eine Schwangerschaft eintritt, muss die Patientin auf das teratogene Risiko einer Therapie mit REBETOL 200 mg für den Fötus hingewiesen werden. Patientinnen oder Partner von Patientinnen sollten jede Schwangerschaft, die während der Behandlung oder innerhalb von 6 Monaten nach Beendigung der Behandlung auftritt, unverzüglich ihrem Arzt melden. Verschreibende Ärzte sollten solche Fälle telefonisch unter 1-800-593-2214 melden.
Risiken versus Vorteile
Patienten, die REBETOL-Kapseln erhalten, sollten über Nutzen und Risiken der Behandlung aufgeklärt, über die angemessene Anwendung informiert und an den Patienten überwiesen werden Leitfaden für Medikamente . Die Patienten sollten darüber informiert werden, dass die Auswirkungen der Behandlung einer Hepatitis-C-Infektion auf die Übertragung nicht bekannt sind und dass geeignete Vorsichtsmaßnahmen getroffen werden sollten, um eine Übertragung des Hepatitis-C-Virus zu verhindern.
Die Patienten sollten darüber informiert werden, was zu tun ist, falls sie eine REBETOL-Dosis vergessen; die vergessene Dosis sollte so bald wie möglich noch am selben Tag eingenommen werden. Patienten sollten die nächste Dosis nicht verdoppeln. Patienten sollten angewiesen werden, sich bei Fragen an ihren Arzt zu wenden.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Ribavirin verursachte keine Zunahme bei irgendeinem Tumortyp, wenn es über 6 Monate im Mausmodell mit transgenem p53-Mangel in Dosen von bis zu 300 mg/kg verabreicht wurde (geschätztes menschliches Äquivalent von 25 mg/kg, basierend auf der Anpassung der Körperoberfläche für einen 60 kg schweren Erwachsenen). ; etwa das 1,9-fache der maximal empfohlenen Tagesdosis für den Menschen). Ribavirin war nicht karzinogen, wenn es Ratten über 2 Jahre in Dosen von bis zu 40 mg/kg verabreicht wurde (geschätztes menschliches Äquivalent von 5,71 mg/kg, basierend auf der Anpassung der Körperoberfläche für einen 60 kg schweren Erwachsenen).
Mutagenese
Ribavirin zeigte in mehreren Genotoxizitätsassays ein erhöhtes Auftreten von Mutationen und Zelltransformationen. Ribavirin war im Balb/3T3-In-vitro-Zelltransformationsassay aktiv. Mutagene Aktivität wurde im Maus-Lymphom-Assay und bei Dosen von 20 bis 200 mg/kg (geschätztes menschliches Äquivalent von 1,67 bis 16,7 mg/kg, basierend auf der Anpassung der Körperoberfläche für einen 60 kg schweren Erwachsenen; 0,1- bis 1-faches des Maximums) beobachtet empfohlene menschliche 24-Stunden-Dosis von Ribavirin) in einem Maus-Mikronukleus-Assay. Ein Dominant-Letal-Assay bei Ratten war negativ, was darauf hinweist, dass Mutationen, die bei Ratten auftraten, nicht durch männliche Gameten übertragen wurden.
Beeinträchtigung der Fruchtbarkeit
Ribavirin zeigte signifikante embryozide und teratogene Wirkungen bei Dosen, die deutlich unter der empfohlenen Humandosis bei allen Tierarten lagen, an denen angemessene Studien durchgeführt wurden. Fehlbildungen des Schädels, des Gaumens, des Auges, des Kiefers, der Gliedmaßen, des Skeletts und des Gastrointestinaltrakts wurden festgestellt. Die Inzidenz und Schwere teratogener Wirkungen nahmen mit steigender Dosis des Arzneimittels zu. Das Überleben von Föten und Nachkommen war reduziert. In konventionellen Embryotoxizitäts-/Teratogenitätsstudien an Ratten und Kaninchen lagen die beobachteten Dosen ohne Wirkung weit unter denen für die vorgeschlagene klinische Anwendung (0,3 mg/kg/Tag sowohl für Ratten als auch für Kaninchen; etwa das 0,06-Fache der empfohlenen 24-Stunden-Menschdosis von Ribavirin). In einer peri-/postnatalen Toxizitätsstudie an Ratten, denen bis zu 1 mg/kg/Tag oral verabreicht wurde (geschätzte menschliche Äquivalentdosis von 0,17 mg/kg, basierend auf der Anpassung der Körperoberfläche für einen 60 kg schweren Erwachsenen), wurden keine maternale Toxizität oder Auswirkungen auf die Nachkommen beobachtet ; ungefähr das 0,01-fache der maximal empfohlenen 24-Stunden-Dosis von Ribavirin für den Menschen) [siehe KONTRAINDIKATIONEN , und WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Gebärfähige Frauen und Partner von gebärfähigen Frauen sollten REBETOL nicht erhalten, es sei denn, die Patientin und ihr Partner wenden eine wirksame Empfängnisverhütung an (zwei zuverlässige Formen). Basierend auf einer Mehrfachdosis-Halbwertszeit (t1/2) von Ribavirin von 12 Tagen muss eine wirksame Empfängnisverhütung für 6 Monate nach der Therapie angewendet werden (z. B. 15 Halbwertszeiten der Clearance für Ribavirin).
REBETOL sollte bei gebärfähigen Männern mit Vorsicht angewendet werden. In Studien an Mäusen zur Bewertung des zeitlichen Verlaufs und der Reversibilität der Ribavirin-induzierten Hodendegeneration bei Dosen von 15 bis 150 mg/kg/Tag (geschätztes Humanäquivalent von 1,25 bis 12,5 mg/kg/Tag, basierend auf der Anpassung der Körperoberfläche an a 60-kg-Erwachsener; das 0,1- bis 0,8-fache der maximalen menschlichen 24-Stunden-Dosis von Ribavirin) über 3 oder 6 Monate verabreicht, traten Anomalien in den Spermien auf. Nach Beendigung der Behandlung war innerhalb von 1 oder 2 Spermatogenesezyklen im Wesentlichen eine vollständige Genesung von der Ribavirin-induzierten testikulären Toxizität erkennbar.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschaftskategorie X
[Sehen KONTRAINDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , und Nichtklinische Toxikologie ].
Behandlung und Nachbehandlung:
Potentielles Risiko für den Fötus
Es ist bekannt, dass sich Ribavirin in intrazellulären Komponenten anreichert, aus denen es sehr langsam ausgeschieden wird. Es ist nicht bekannt, ob Ribavirin, das in Spermien enthalten ist, eine potenzielle teratogene Wirkung auf die Befruchtung der Eizellen ausübt. In einer Studie an Ratten wurde der Schluss gezogen, dass Ribavirin bei Dosen von bis zu 200 mg/kg über 5 Tage (geschätzte humanäquivalente Dosen von 7,14 bis 28,6 mg/kg, basierend auf einer Anpassung der Körperoberfläche für einen 60 kg Erwachsener; bis zum 1,7-Fachen der maximal empfohlenen Ribavirin-Dosis für den Menschen). Aufgrund der möglichen humanteratogenen Wirkungen von Ribavirin sollten männliche Patienten jedoch angewiesen werden, alle Vorsichtsmaßnahmen zu treffen, um das Risiko einer Schwangerschaft für ihre Partnerinnen zu vermeiden.
Frauen im gebärfähigen Alter sollten REBETOL nicht erhalten, es sei denn, sie wenden während des Therapiezeitraums eine wirksame Empfängnisverhütung (zwei zuverlässige Formen) an. Darüber hinaus sollte für 6 Monate nach der Therapie eine wirksame Verhütungsmethode angewendet werden, basierend auf einer Mehrfachdosis-Halbwertszeit (t1/2) von Ribavirin von 12 Tagen.
Männliche Patienten und ihre Partnerinnen müssen während der Behandlung mit REBETOL und für die 6-monatige Nachbehandlungszeit (z. B. 15 Halbwertszeiten für die Clearance von Ribavirin aus dem Körper) eine wirksame Verhütung (zwei zuverlässige Formen) praktizieren.
Ein Ribavirin-Schwangerschaftsregister wurde eingerichtet, um den mütterlich-fötalen Ausgang von Schwangerschaften bei weiblichen Patienten und weiblichen Partnern von männlichen Patienten zu überwachen, die während der Behandlung und für 6 Monate nach Beendigung der Behandlung Ribavirin ausgesetzt waren. Ärzte und Patienten werden ermutigt, solche Fälle telefonisch unter 1-800-593-2214 zu melden.
Stillende Mutter
Es ist nicht bekannt, ob das Produkt REBETOL in die Muttermilch übergeht. Aufgrund der Möglichkeit schwerwiegender Nebenwirkungen des Arzneimittels bei gestillten Säuglingen sollte eine Entscheidung getroffen werden, ob das Stillen abgebrochen oder REBETOL verschoben oder abgesetzt werden soll.
Pädiatrische Verwendung
Die Sicherheit und Wirksamkeit von REBETOL 200 mg in Kombination mit PegIntron wurde bei pädiatrischen Patienten unter 3 Jahren nicht nachgewiesen. Bei der Behandlung mit REBETOL/INTRON A sollten bei der Entscheidung, einen pädiatrischen Patienten zu behandeln, Anzeichen einer Krankheitsprogression, wie z. B. Leberentzündung und Fibrose, sowie prognostische Faktoren für das Ansprechen, HCV-Genotyp und Viruslast berücksichtigt werden. Der Nutzen der Behandlung sollte gegen die beobachteten Sicherheitsbefunde abgewogen werden.
Langzeit-Follow-up-Daten bei pädiatrischen Probanden weisen darauf hin, dass REBETOL in Kombination mit PegIntron oder mit INTRON A eine Wachstumshemmung hervorrufen kann, die bei einigen Patienten zu einer verringerten Körpergröße führt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Suizidgedanken oder -versuche traten bei pädiatrischen Patienten, vor allem Jugendlichen, häufiger auf als bei erwachsenen Patienten (2,4 % vs. 1 %) während der Behandlung und während der Nachbeobachtung außerhalb der Therapie [sehen WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Wie bei erwachsenen Patienten erlebten pädiatrische Patienten andere psychiatrische Nebenwirkungen (z. B. Depression, emotionale Labilität, Somnolenz), Anämie und Neutropenie [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Geriatrische Verwendung
Klinische Studien zur Therapie mit REBETOL/INTRON A oder PegIntron schlossen keine ausreichende Anzahl von Probanden ab 65 Jahren ein, um festzustellen, ob sie anders ansprechen als jüngere Probanden.
Es ist bekannt, dass REBETOL 200 mg im Wesentlichen über die Nieren ausgeschieden wird, und das Risiko toxischer Reaktionen auf dieses Arzneimittel kann bei Patienten mit eingeschränkter Nierenfunktion größer sein. Da ältere Patienten häufig eine eingeschränkte Nierenfunktion haben, sollte die Dosis sorgfältig ausgewählt werden. Die Nierenfunktion sollte überwacht und die Dosis entsprechend angepasst werden. REBETOL 200 mg sollte nicht bei Patienten mit einer Kreatinin-Clearance von weniger als 50 ml/min angewendet werden [siehe KONTRAINDIKATIONEN ].
Im Allgemeinen sollten REBETOL 200 mg Kapseln älteren Patienten mit Vorsicht verabreicht werden, beginnend am unteren Ende des Dosierungsbereichs, um die größere Häufigkeit von verminderter Leber- und Herzfunktion und von Begleiterkrankungen oder anderen medikamentösen Therapien widerzuspiegeln. In klinischen Studien hatten ältere Patienten häufiger Anämie (67 %) als jüngere Patienten (28 %) [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Empfänger von Organtransplantationen
Die Sicherheit und Wirksamkeit von INTRON A und PegIntron allein oder in Kombination mit REBETOL zur Behandlung von Hepatitis C bei Empfängern von Leber- oder anderen Organtransplantaten wurde nicht nachgewiesen. In einer kleinen (n=16) monozentrischen, unkontrollierten Fallerfahrung trat Nierenversagen bei Empfängern von Nierentransplantaten, die eine Kombinationstherapie mit Interferon alpha und Ribavirin erhielten, häufiger auf als aufgrund der früheren Erfahrungen des Zentrums mit Empfängern von Nierentransplantaten, die keine Kombinationstherapie erhielten, erwartet wurde. Die Beziehung des Nierenversagens zur Abstoßung des Nierentransplantats ist nicht klar.
HIV- oder HBV-Koinfektion
Die Sicherheit und Wirksamkeit von PegIntron/REBETOL 200 mg und INTRON A/REBETOL zur Behandlung von Patienten mit HCV-Koinfektion mit HIV oder HBV wurden nicht nachgewiesen.
ÜBERDOSIS
Es liegen nur begrenzte Erfahrungen mit Überdosierung vor. Akute Einnahme von bis zu 20 g REBETOL-Kapseln, Einnahme von INTRON A von bis zu 120 Millionen Einheiten und subkutane Dosen von INTRON A bis zum 10-Fachen der empfohlenen Dosis wurden berichtet. Die beobachteten primären Wirkungen sind eine erhöhte Inzidenz und Schwere der Nebenwirkungen im Zusammenhang mit der therapeutischen Anwendung von INTRON A und REBETOL. Es wurde jedoch über Anomalien der Leberenzyme, Nierenversagen, Blutungen und Myokardinfarkt bei Verabreichung von subkutanen Einzeldosen von INTRON A berichtet, die die Dosierungsempfehlungen überschritten.
Es gibt kein spezifisches Antidot für eine Überdosierung von INTRON A oder REBETOL 200 mg, und Hämodialyse und Peritonealdialyse sind zur Behandlung einer Überdosierung dieser Mittel nicht wirksam.
KONTRAINDIKATIONEN
Die REBETOL-Kombinationstherapie ist kontraindiziert bei:
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Ribavirin ist ein antivirales Mittel [siehe Mikrobiologie ].
Pharmakokinetik
Die pharmakokinetischen Eigenschaften nach Einzel- und Mehrfachdosis bei Erwachsenen sind in Tabelle 11 zusammengefasst. Ribavirin wurde nach oraler Verabreichung schnell und umfassend resorbiert. Aufgrund des First-Pass-Metabolismus betrug die absolute Bioverfügbarkeit jedoch durchschnittlich 64 % (44 %). Nach Einzeldosen von 200 bis 1200 mg Ribavirin bestand eine lineare Beziehung zwischen Dosis und AUCtf (AUC vom Zeitpunkt Null bis zur letzten messbaren Konzentration). Die Beziehung zwischen Dosis und Cmax war krummlinig und neigte dazu, oberhalb von Einzeldosen von 400 bis 600 mg asymptotisch zu werden.
Nach mehrfacher oraler Gabe, basierend auf AUC12h, wurde eine 6-fache Akkumulation von Ribavirin im Plasma beobachtet. Nach oraler Gabe von 600 mg zweimal täglich wurde der Steady-State nach etwa 4 Wochen erreicht, mit mittleren Steady-State-Plasmakonzentrationen von 2200 ng/ml (37 %). Nach Absetzen der Dosierung betrug die mittlere Halbwertszeit 298 (30 %) Stunden, was wahrscheinlich auf eine langsame Elimination aus Nichtplasmakompartimenten zurückzuführen ist.
Wirkung von Antazida auf die Absorption von Ribavirin
Die gleichzeitige Verabreichung von REBETOL-Kapseln mit einem Antazida, das Magnesium, Aluminium und Simethicon enthält, führte zu einer 14 %igen Abnahme der mittleren AUCtf von Ribavirin. Die klinische Relevanz der Ergebnisse dieser Einzeldosisstudie ist nicht bekannt.
Gewebeverteilung
Der Transport von Ribavirin in Kompartimente außerhalb des Plasmas wurde am ausführlichsten in roten Blutkörperchen untersucht, und es wurde festgestellt, dass er hauptsächlich über einen äquilibrierenden Nukleosidtransporter vom es-Typ erfolgt. Dieser Transportertyp ist auf praktisch allen Zelltypen vorhanden und kann für das umfangreiche Verteilungsvolumen verantwortlich sein. Ribavirin bindet nicht an Plasmaproteine.
Stoffwechsel und Ausscheidung
Ribavirin hat zwei Metabolisierungswege: (i) einen reversiblen Phosphorylierungsweg in kernhaltigen Zellen; und (ii) ein Abbauweg, der eine Deribosylierung und eine Amidhydrolyse umfasst, um einen Triazolcarbonsäure-Metaboliten zu ergeben. Ribavirin und seine Metaboliten Triazolcarboxamid und Triazolcarbonsäure werden renal ausgeschieden. Nach oraler Gabe von 600 mg 14C-Ribavirin wurden innerhalb von 336 Stunden etwa 61 % bzw. 12 % der Radioaktivität im Urin bzw. im Stuhl ausgeschieden. Unverändertes Ribavirin machte 17 % der verabreichten Dosis aus.
Besondere Populationen
Nierenfunktionsstörung
Die Pharmakokinetik von Ribavirin wurde nach Verabreichung einer oralen Einzeldosis (400 mg) Ribavirin an nicht HCV-infizierte Probanden mit Nierenfunktionsstörungen unterschiedlichen Grades untersucht. Der mittlere AUCtf-Wert war bei Probanden mit Kreatinin-Clearance-Werten zwischen 10 und 30 ml/min dreimal höher als bei Kontrollpersonen (Kreatinin-Clearance größer als 90 ml/min). Bei Probanden mit Kreatinin-Clearance-Werten zwischen 30 und 60 ml/min war die AUCtf im Vergleich zu Kontrollprobanden doppelt so hoch. Die erhöhte AUCtf scheint auf eine Verringerung der renalen und nicht-renalen Clearance bei diesen Patienten zurückzuführen zu sein. Die Wirksamkeitsstudien der Phase 3 schlossen Probanden mit Kreatinin-Clearance-Werten über 50 ml/min ein. Die Pharmakokinetik von Ribavirin nach Mehrfachgabe kann bei Patienten mit Nierenfunktionsstörung nicht genau vorhergesagt werden. Ribavirin wird durch Hämodialyse nicht wirksam entfernt.
Patienten mit einer Kreatinin-Clearance von weniger als 50 ml/min sollten nicht mit REBETOL behandelt werden [siehe KONTRAINDIKATIONEN ].
Leberfunktionsstörung
Die Auswirkung einer Leberfunktionsstörung wurde nach einer oralen Einzeldosis von Ribavirin (600 mg) beurteilt. Die mittleren AUCtf-Werte waren bei Patienten mit leichter, mittelschwerer oder schwerer Leberfunktionsstörung (Child-Pugh-Klassifikation A, B oder C) im Vergleich zu Kontrollpersonen nicht signifikant unterschiedlich. Die mittleren Cmax-Werte stiegen jedoch mit der Schwere der Leberfunktionsstörung und waren bei Patienten mit schwerer Leberfunktionsstörung doppelt so hoch wie bei Kontrollpersonen.
Ältere Patienten
Pharmakokinetische Untersuchungen bei älteren Patienten wurden nicht durchgeführt.
Geschlecht
In einer Einzeldosisstudie mit 18 männlichen und 18 weiblichen Probanden wurden keine klinisch signifikanten pharmakokinetischen Unterschiede festgestellt.
Pädiatrische Patienten
Die pharmakokinetischen Eigenschaften von REBETOL 200 mg Kapseln und INTRON A nach Mehrfachgabe bei Kindern und Jugendlichen mit chronischer Hepatitis C im Alter zwischen 5 und 16 Jahren sind in Tabelle 12 zusammengefasst pädiatrische Fächer.
Die vollständigen pharmakokinetischen Eigenschaften von REBETOL Lösung zum Einnehmen wurden bei Kindern und Jugendlichen nicht bestimmt. Die Cmin-Werte von Ribavirin waren nach Verabreichung von REBETOL Lösung zum Einnehmen oder REBETOL 200 mg Kapseln während einer 48-wöchigen Therapie bei pädiatrischen Patienten (3 bis 16 Jahre) ähnlich.
Es wurde eine klinische Studie mit Kindern und Jugendlichen mit chronischer Hepatitis C im Alter zwischen 3 und 17 Jahren durchgeführt, in der die Pharmakokinetik von PegIntron und REBETOL (Kapseln und Lösung zum Einnehmen) bewertet wurde. Bei pädiatrischen Probanden, die eine an die Körperoberfläche angepasste Dosierung von PegIntron von 60 mcg/m²/Woche erhielten, wurde die logarithmisch transformierte Ratio-Schätzung der Exposition während des Dosierungsintervalls auf 58 % [90 %-KI: 141 %, 177 %] höher als prognostiziert bei Erwachsenen beobachtet, die 1,5 mcg/kg/Woche erhielten. Die Pharmakokinetik von REBETOL (dosisnormalisiert) in dieser Studie war ähnlich wie in einer früheren Studie mit REBETOL 200 mg in Kombination mit INTRON A bei pädiatrischen Probanden und Erwachsenen.
Wirkung von Nahrung auf die Resorption von Ribavirin
Sowohl AUCtf als auch Cmax erhöhten sich um 70 %, wenn REBETOL-Kapseln mit einer fettreichen Mahlzeit (841 kcal, 53,8 g Fett, 31,6 g Protein und 57,4 g Kohlenhydrate) in einer pharmakokinetischen Studie mit Einzeldosis verabreicht wurden [siehe DOSIERUNG UND ANWENDUNG ].
Mikrobiologie
Wirkmechanismus
Der Mechanismus, durch den Ribavirin zu seiner antiviralen Wirksamkeit in der Klinik beiträgt, ist noch nicht vollständig geklärt. Ribavirin hat in Gewebekultur eine direkte antivirale Aktivität gegen viele RNA-Viren. Ribavirin erhöht die Mutationshäufigkeit im Genom mehrerer Viren und Ribavirintriphosphat hemmt die HCV-Polymerase in einer biochemischen Reaktion.
Antivirale Aktivität in der Zellkultur
Die antivirale Aktivität von Ribavirin im HCV-Replikon ist nicht gut verstanden und wurde aufgrund der zellulären Toxizität von Ribavirin nicht definiert. In Gewebekulturen anderer RNA-Viren wurde eine direkte antivirale Aktivität beobachtet. Die Anti-HCV-Aktivität von Interferon wurde in Zellen nachgewiesen, die selbstreplizierende HCV-RNS (HCV-Replikonzellen) oder eine HCV-Infektion enthielten.
Widerstand
HCV-Genotypen zeigen eine große Variabilität in ihrer Reaktion auf eine Therapie mit pegyliertem rekombinantem humanem Interferon/Ribavirin. Genetische Veränderungen im Zusammenhang mit der variablen Reaktion wurden nicht identifiziert.
Kreuzresistenz
Es wurde keine Kreuzresistenz zwischen pegylierten/nicht-pegylierten Interferonen und Ribavirin berichtet.
Tiertoxikologie und Pharmakologie
Langzeitstudien an Maus und Ratte [18 bis 24 Monate; Dosen von 20 bis 75 bzw. 10 bis 40 mg/kg/Tag (geschätzte menschliche Äquivalentdosen von 1,67 bis 6,25 bzw. 1,43 bis 5,71 mg/kg/Tag, basierend auf der Anpassung der Körperoberfläche für einen 60 kg schweren Erwachsenen; ca 0,1- bis 0,4-fache der maximalen menschlichen 24-Stunden-Dosis von Ribavirin)] haben einen Zusammenhang zwischen einer chronischen Ribavirin-Exposition und einem erhöhten Auftreten von Gefäßläsionen (Mikroblutungen) bei Mäusen gezeigt. Bei Ratten trat bei Kontrollen eine Netzhautdegeneration auf, aber die Inzidenz war bei mit Ribavirin behandelten Ratten erhöht.
In einer Studie, in der Rattenjungen postnatal Ribavirin in Dosen von 10, 25 und 50 mg/kg/Tag verabreicht wurde, traten arzneimittelbedingte Todesfälle bei 50 mg/kg auf (bei Rattenjungen-Plasmakonzentrationen unterhalb der menschlichen Plasmakonzentrationen beim Menschen). therapeutische Dosis) zwischen den Studientagen 13 und 48. Rattenwelpen, denen von den postnatalen Tagen 7 bis 63 eine Dosis verabreicht wurde, zeigten bei allen Dosen eine geringfügige, dosisabhängige Abnahme des Gesamtwachstums, die sich anschließend als leichte Abnahme des Körpergewichts, der Scheitel-Steiß-Länge, und Knochenlänge. Diese Wirkungen zeigten Hinweise auf Reversibilität, und es wurden keine histopathologischen Wirkungen auf den Knochen beobachtet. Es wurden keine Auswirkungen von Ribavirin in Bezug auf das neurologische Verhalten oder die reproduktive Entwicklung beobachtet.
Klinische Studien
Klinische Studie 1 bewertete die PegIntron-Monotherapie. Sehen PegIntron-Kennzeichnung für Informationen zu dieser Studie.
REBETOL/PegIntron-Kombinationstherapie
Erwachsene Themen
Studie 2
Eine randomisierte Studie verglich die Behandlung mit zwei PegIntron/REBETOL 200 mg-Schemata [PegIntron 1,5 µg/kg subkutan einmal wöchentlich/REBETOL 800 mg oral täglich (aufgeteilt auf mehrere Dosen); PegIntron 1,5 µg/kg subkutan einmal wöchentlich für 4 Wochen, dann 0,5 µg/kg subkutan einmal wöchentlich für 44 Wochen/REBETOL 1000 oder 1200 mg oral täglich (in aufgeteilten Dosen)] mit INTRON A [3 Mio. I.E. subkutan dreimal wöchentlich/REBETOL 1000 oder 1200 mg p.o. täglich (in aufgeteilten Dosen)] bei 1530 Erwachsenen mit chronischer Hepatitis C. Interferon-naive Patienten wurden 48 Wochen lang behandelt und 24 Wochen nach der Behandlung nachbeobachtet. Geeignete Probanden hatten eine kompensierte Lebererkrankung, nachweisbare HCV-RNA, erhöhte ALT und eine mit chronischer Hepatitis übereinstimmende Leberhistopathologie.
Das Ansprechen auf die Behandlung wurde als nicht nachweisbare HCV-RNA 24 Wochen nach der Behandlung definiert (siehe Tabelle 13). Die Ansprechrate auf die Dosis von PegIntron 1,5 µg/kg und Ribavirin 800 mg war höher als die Ansprechrate auf INTRON A/REBETOL (siehe Tabelle 13). Die Ansprechrate auf PegIntron 1,5→0,5 µg/kg/REBETOL 200 mg war im Wesentlichen gleich als Antwort auf INTRON A/REBETOL (Daten nicht gezeigt).
Patienten mit viralem Genotyp 1 zeigten unabhängig von der Viruslast eine niedrigere Ansprechrate auf PegIntron (1,5 µg/kg)/REBETOL (800 mg) im Vergleich zu Patienten mit anderen viralen Genotypen. Patienten mit beiden schlechten Prognosefaktoren (Genotyp 1 und hohe Viruslast) hatten eine Ansprechrate von 30 % (78/256) im Vergleich zu einer Ansprechrate von 29 % (71/247) bei der INTRON A/REBETOL-Kombinationstherapie.
Bei Personen mit geringerem Körpergewicht traten tendenziell höhere Nebenwirkungsraten auf [vgl NEBENWIRKUNGEN und höhere Ansprechraten als Probanden mit höherem Körpergewicht. Die Unterschiede in den Ansprechraten zwischen den Behandlungsarmen variierten nicht wesentlich mit dem Körpergewicht.
Die Ansprechraten auf die Behandlung mit der PegIntron/REBETOL-Kombinationstherapie betrugen 49 % bei Männern und 56 % bei Frauen. Die Rücklaufquoten waren bei Afroamerikanern und Hispanoamerikanern niedriger und bei Asiaten höher als bei Kaukasiern. Obwohl Afroamerikaner im Vergleich zu Kaukasiern einen höheren Anteil an schlechten Prognosefaktoren aufwiesen, reichte die Anzahl der untersuchten Nicht-Kaukasier (11 % der Gesamtzahl) nicht aus, um aussagekräftige Schlussfolgerungen über Unterschiede in den Ansprechraten nach Berücksichtigung prognostischer Faktoren in dieser Studie zu ziehen.
Bei 68 % der Patienten wurden vor und nach der Behandlung Leberbiopsien entnommen. Im Vergleich zum Ausgangswert wurde bei etwa zwei Dritteln der Patienten in allen Behandlungsgruppen eine leichte Verringerung der Entzündung beobachtet.
Studie 3
In einer großen gemeinschaftsbasierten Studie in den Vereinigten Staaten wurden 4913 Patienten mit chronischer Hepatitis C randomisiert, um einmal wöchentlich PegIntron 1,5 µg/kg subkutan in Kombination mit einer Dosis von 800 bis 1400 mg REBETOL 200 mg (gewichtsbasierte Dosierung [WBD]) oder zu erhalten 800 mg (pauschal) oral täglich (in aufgeteilten Dosen) für 24 oder 48 Wochen, je nach Genotyp. Das Ansprechen auf die Behandlung wurde als nicht nachweisbare HCV-RNA (basierend auf einem Assay mit einer unteren Nachweisgrenze von 125 IE/ml) 24 Wochen nach der Behandlung definiert.
Die Behandlung mit PegIntron 1,5 µg/kg und REBETOL 800 bis 1400 mg führte zu einem stärkeren anhaltenden virologischen Ansprechen im Vergleich zu PegIntron in Kombination mit einer pauschalen Tagesdosis von 800 mg REBETOL. Personen mit einem Gewicht von mehr als 105 kg erzielten den größten Nutzen mit WBD, obwohl auch bei Personen mit einem Gewicht von mehr als 85 bis 105 kg ein bescheidener Nutzen beobachtet wurde (siehe Tabelle 14). Der Nutzen von WBD bei Probanden mit einem Gewicht von mehr als 85 kg wurde mit den HCV-Genotypen 1–3 beobachtet. Es waren keine ausreichenden Daten verfügbar, um Schlussfolgerungen zu anderen Genotypen zu ziehen. Die Anwendung von WBD führte zu einer erhöhten Inzidenz von Anämie [siehe NEBENWIRKUNGEN ].
Insgesamt 1552 Patienten mit einem Gewicht von mehr als 65 kg in Studie 3 hatten Genotyp 2 oder 3 und wurden randomisiert einer 24- oder 48-wöchigen Therapie zugeteilt. Bei der längeren Behandlungsdauer wurde kein zusätzlicher Nutzen beobachtet.
Studie 4
Eine große randomisierte Studie verglich die Sicherheit und Wirksamkeit einer Behandlung über 48 Wochen mit zwei PegIntron/REBETOL 200 mg-Schemata [PegIntron 1,5 µg/kg und 1 µg/kg subkutan einmal wöchentlich jeweils in Kombination mit REBETOL 800 bis 1400 mg p.o. täglich (in zwei geteilte Dosen). Dosen)] und Pegasys 180 µg subkutan einmal wöchentlich in Kombination mit Copegus 1000 bis 1200 mg p.o. täglich (in zwei getrennten Dosen) bei 3070 nicht vorbehandelten Erwachsenen mit chronischer Hepatitis C vom Genotyp 1 HCV-RNA oder größer oder gleich 2 log10 Reduktion gegenüber dem Ausgangswert) nach Behandlung Woche 12 war das Kriterium für den Abbruch der Behandlung. SVR wurde definiert als nicht nachweisbare HCV-RNA (Roche COBAS TaqMan Assay, eine untere Bestimmungsgrenze von 27 IE/ml) 24 Wochen nach der Behandlung (siehe Tabelle 15).
Die SVR-Gesamtraten waren in den drei Behandlungsgruppen ähnlich. Unabhängig von der Behandlungsgruppe waren die SVR-Raten bei Patienten mit schlechten Prognosefaktoren niedriger. Patienten mit schlechten Prognosefaktoren, die auf PegIntron (1,5 µg/kg)/REBETOL oder Pegasys/Copegus randomisiert wurden, erreichten jedoch höhere SVR-Raten im Vergleich zu ähnlichen Patienten, die auf PegIntron 1 µg/kg/REBETOL randomisiert wurden. Für PegIntron 1,5 mcg/kg und die REBETOL-Dosis waren die SVR-Raten für Patienten mit und ohne die folgenden Prognosefaktoren wie folgt: Zirrhose (10 % vs. 42 %), normale ALT-Spiegel (32 % vs. 42 %), virale Ausgangswerte Last größer als 600.000 IE/ml (35 % vs. 61 %), 40 Jahre und älter (38 % vs. 50 %) und Afroamerikaner (23 % vs. 44 %). Bei Patienten mit nicht nachweisbarer HCV-RNA in Behandlungswoche 12, die PegIntron (1,5 mcg/kg)/REBETOL 200 mg erhielten, betrug die SVR-Rate 81 % (328/407).
Studie 5 – REBETOL/PegIntron-Kombinationstherapie bei vorherigem Behandlungsversagen
In einer nicht vergleichenden Studie wurden 2293 Patienten mit mittelschwerer bis schwerer Fibrose, bei denen eine vorherige Behandlung mit einer Kombination aus alpha-Interferon/Ribavirin fehlschlug, erneut mit PegIntron, 1,5 µg/kg subkutan, einmal wöchentlich, in Kombination mit gewichtsadaptiertem Ribavirin behandelt. Zu den geeigneten Probanden gehörten frühere Nonresponder (Probanden, die am Ende einer mindestens 12-wöchigen Behandlung HCV-RNA-positiv waren) und frühere Rückfälle (Probanden, die am Ende einer mindestens 12-wöchigen Behandlung HCVRNA-negativ waren und anschließend nach der Behandlung einen Rückfall erlitten nachverfolgen). Patienten, die in Woche 12 negativ waren, wurden 48 Wochen lang behandelt und 24 Wochen nach der Behandlung nachbeobachtet. Das Ansprechen auf die Behandlung wurde definiert als nicht nachweisbare HCV-RNA 24 Wochen nach der Behandlung (gemessen mit einem forschungsbasierten Test, Nachweisgrenze 125 IE/ml). Die Gesamtansprechrate betrug 22 % (497/2293) (99 % KI: 19,5; 23,9). Patienten mit den folgenden Merkmalen profitierten mit geringerer Wahrscheinlichkeit von einer erneuten Behandlung: vorheriges Nichtansprechen, vorherige Behandlung mit pegyliertem Interferon, signifikante überbrückende Fibrose oder Zirrhose und Infektion vom Genotyp 1.
Die Raten des anhaltenden virologischen Ansprechens auf eine erneute Behandlung nach Merkmalen zu Studienbeginn sind in Tabelle 16 zusammengefasst.
Das Erreichen einer nicht nachweisbaren HCV-RNA in Behandlungswoche 12 war ein starker Prädiktor für SVR. In dieser Studie erreichten 1470 (64 %) Patienten in Behandlungswoche 12 keine nicht nachweisbare HCV-RNA und ihnen wurde aufgrund eines unzureichenden Ansprechens auf die Behandlung die Aufnahme in Langzeitbehandlungsstudien angeboten. Von den 823 (36 %) Patienten, bei denen in Behandlungswoche 12 keine HCV-RNA nachweisbar war, hatten diejenigen, die mit Genotyp 1 infiziert waren, eine SVR von 48 % (245/507), mit einer Bandbreite von Reaktionen nach Fibrose-Scores (F4-F2) von 39-55%. Mit Genotyp 2/3 infizierte Patienten, bei denen in Behandlungswoche 12 keine HCV-RNA nachweisbar war, hatten eine Gesamt-SVR von 70 % (196/281) mit einem Ansprechbereich nach Fibrose-Scores (F4-F2) von 60-83 %. Bei allen Genotypen waren höhere Fibrose-Scores mit einer geringeren Wahrscheinlichkeit verbunden, eine SVR zu erreichen.
Pädiatrische Fächer
Zuvor unbehandelte pädiatrische Probanden im Alter von 3 bis 17 Jahren mit kompensierter chronischer Hepatitis C und nachweisbarer HCV-RNA wurden mit REBETOL 15 mg/kg pro Tag und PegIntron 60 mcg/m² einmal wöchentlich für 24 oder 48 Wochen behandelt, basierend auf dem HCV-Genotyp und dem viralen Ausgangswert Belastung. Alle Probanden sollten nach der Behandlung 24 Wochen lang nachbeobachtet werden. Insgesamt wurden 107 Patienten behandelt, von denen 52 % weiblich, 89 % Kaukasier und 67 % mit HCV-Genotyp 1 infiziert waren. Patienten, die mit den Genotypen 1, 4 oder Genotyp 3 mit HCV-RNA größer oder gleich 600.000 infiziert waren IE/ml erhielten eine 48-wöchige Therapie, während diejenigen, die mit Genotyp 2 oder Genotyp 3 mit einer HCV-RNA von weniger als 600.000 IE/ml infiziert waren, eine 24-wöchige Therapie erhielten. Die Versuchsergebnisse sind in Tabelle 17 zusammengefasst.
REBETOL/INTRON Eine Kombinationstherapie
Erwachsene Themen
Zuvor unbehandelte Probanden
Erwachsene mit kompensierter chronischer Hepatitis C und nachweisbarer HCV-RNA (bewertet von einem Zentrallabor unter Verwendung eines forschungsbasierten RT-PCR-Assays), die zuvor nicht mit Alpha-Interferon behandelt worden waren, wurden in zwei multizentrische, doppelblinde Studien (USA und international) aufgenommen. und randomisiert, um REBETOL 200 mg Kapseln 1200 mg/Tag (1000 mg/Tag für Personen mit einem Gewicht von weniger als oder gleich 75 kg) und INTRON A 3 Mio. E. dreimal wöchentlich oder INTRON A und Placebo für 24 oder 48 Wochen gefolgt von 24 Wochen zu erhalten Nachsorge außerhalb der Therapie. Die internationale Studie enthielt keinen 24-wöchigen Behandlungsarm mit INTRON A und Placebo. In die US-Studie wurden 912 Probanden aufgenommen, die zu Studienbeginn zu 67 % männlich, zu 89 % Kaukasier mit einem mittleren Knodell-HAI-Score (I+II+III) von 7,5 und zu 72 % vom Genotyp 1 waren. Die internationale Studie wurde in Europa, Israel, durchgeführt , Kanada und Australien, nahmen 799 Probanden (65 % männlich, 95 % Kaukasier, mittlerer Knodell-Score 6,8 und 58 % Genotyp 1) auf.
Die Versuchsergebnisse sind in Tabelle 18 zusammengefasst.
Von den Patienten, die bis Woche 24 der Behandlung mit REBETOL/INTRON A keine HCV-RNA unterhalb der Nachweisgrenze des forschungsbasierten Assays erreicht hatten, sprachen weniger als 5 % auf eine zusätzliche 24-wöchige Kombinationsbehandlung an.
Unter den Patienten mit HCV-Genotyp 1, die mit der REBETOL/INTRON A-Therapie behandelt wurden und die nach 24 Wochen HCV-RNA unterhalb der Nachweisgrenze des forschungsbasierten Assays erreichten, zeigten diejenigen, die für eine 48-wöchige Behandlung randomisiert wurden, ein höheres virologisches Ansprechen im Vergleich zu denen in der 24- Woche Behandlungsgruppe. Bei Studienteilnehmern mit HCV-Nicht-Genotyp 1, die für eine REBETOL/INTRON A-Therapie für 48 Wochen im Vergleich zu 24 Wochen randomisiert wurden, wurde kein Anstieg der Ansprechraten beobachtet.
Rückfall-Themen
Patienten mit kompensierter chronischer Hepatitis C und nachweisbarer HCV-RNA (bewertet von einem Zentrallabor unter Verwendung eines forschungsbasierten RT-PCR-Assays), die nach einem oder zwei Interferontherapiezyklen einen Rückfall erlitten hatten (definiert als abnormale Serum-ALT-Spiegel), wurden in zwei aufgenommen multizentrische, doppelblinde Studien (USA und international) und randomisiert, um REBETOL 1200 mg/Tag (1000 mg/Tag für Probanden mit einem Gewicht von ≤ 75 kg) und INTRON A 3 Mio. I.E. dreimal wöchentlich oder INTRON A und Placebo für 24 Wochen gefolgt von zu erhalten 24 Wochen therapiefreie Nachsorge. Die US-Studie umfasste 153 Probanden, die zu Studienbeginn zu 67 % männlich, zu 92 % Kaukasier mit einem mittleren Knodell-HAI-Score (I+II+III) von 6,8 und zu 58 % vom Genotyp 1 waren. Die internationale Studie wurde in Europa, Israel durchgeführt , Kanada und Australien, nahmen 192 Probanden (64 % männlich, 95 % Kaukasier, mittlerer Knodell-Score 6,6 und 56 % Genotyp 1) auf. Die Versuchsergebnisse sind in Tabelle 19 zusammengefasst.
Das virologische und histologische Ansprechen war bei männlichen und weiblichen Probanden sowohl in den zuvor unbehandelten als auch in den Rückfallstudien ähnlich.
Pädiatrische Fächer
Kinder und Jugendliche im Alter von 3 bis 16 Jahren mit kompensierter chronischer Hepatitis C und nachweisbarer HCV-RNA (bewertet von einem Zentrallabor mit einem forschungsbasierten RT-PCR-Assay) wurden mit REBETOL 15 mg/kg pro Tag und INTRON A 3 Mio. I.E./ m² dreimal wöchentlich für 48 Wochen, gefolgt von 24 Wochen therapiefreier Nachsorge. Insgesamt wurden 118 Probanden behandelt, von denen 57 % männlich, 80 % Kaukasier und 78 % vom Genotyp 1 waren. Probanden unter 5 Jahren erhielten REBETOL Lösung zum Einnehmen und Personen ab 5 Jahren erhielten entweder REBETOL Lösung zum Einnehmen oder Kapseln.
Die Versuchsergebnisse sind in Tabelle 20 zusammengefasst.
Patienten mit viralem Genotyp 1 hatten unabhängig von der Viruslast eine geringere Ansprechrate auf die INTRON A/REBETOL-Kombinationstherapie im Vergleich zu Patienten mit Genotyp non-1, 36 % gegenüber 81 %. Patienten mit beiden schlechten Prognosefaktoren (Genotyp 1 und hohe Viruslast) hatten eine Ansprechrate von 26 % (13/50).
INFORMATIONEN ZUM PATIENTEN
Keine Informationen bereitgestellt. Bitte wende dich an die WARNUNGEN UND VORSICHTSMASSNAHMEN Sektion.