Sustiva 200mg, 600mg Efavirenz Verwendung, Nebenwirkungen, Stärke und Dosierung. Preis in Online-Apotheke. Generika medikamente rezeptfrei.
Was ist Sustiva 600 mg und wie wird es angewendet?
Sustiva ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome einer HIV-Infektion. Sustiva kann allein oder mit anderen Medikamenten verwendet werden.
Sustiva gehört zu einer Klasse von Medikamenten namens HIV, NNRTIs.
Es ist nicht bekannt, ob Sustiva bei Kindern unter 3 Monaten sicher und wirksam ist.
Welche Nebenwirkungen kann Sustiva haben?
Sustiva kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Sustiva gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Sustiva. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
SUSTIVA® (Efavirenz) ist ein HIV-1-spezifischer, nicht-nukleosidischer Reverse-Transkriptase-Hemmer (NNRTI). Efavirenz wird chemisch als (S)-6-Chlor-4-(cyclopropylethinyl)-1,4-dihydro4-(trifluormethyl)-2H-3,1-benzoxazin-2-on beschrieben. Seine Summenformel ist C14H9ClF3NO2 und seine Strukturformel ist:
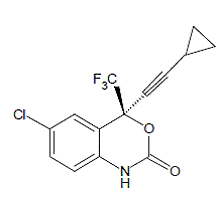
Efavirenz ist ein weißes bis leicht rosa kristallines Pulver mit einer Molekülmasse von 315,68. Es ist in Wasser praktisch unlöslich (
Kapseln
SUSTIVA ist als Kapseln zur oralen Verabreichung mit entweder 50 mg oder 200 mg Efavirenz und den folgenden Hilfsstoffen erhältlich: Lactose-Monohydrat, Magnesiumstearat, Natriumlaurylsulfat und Natriumstärkeglykolat. Die Kapselhülle enthält die folgenden Hilfsstoffe und Farbstoffe: Gelatine, Natriumlaurylsulfat, Titandioxid und/oder gelbes Eisenoxid. Die Kapselhüllen können auch Siliziumdioxid enthalten. Die Kapseln sind mit Tinte bedruckt, die Carmine 40 Blue, FD&C Blue No. 2 und Titandioxid enthält.
Tablets
SUSTIVA 600 mg ist als Filmtablette zur oralen Verabreichung mit 600 mg Efavirenz und den folgenden Hilfsstoffen erhältlich: Croscarmellose-Natrium, Hydroxypropylcellulose, Lactose-Monohydrat, Magnesiumstearat, mikrokristalline Cellulose und Natriumlaurylsulfat. Die Filmbeschichtung enthält Opadry Yellow und Opadry Clear. Die Tabletten sind mit Carnaubawachs poliert und mit violetter Tinte, Opacode WB, bedruckt.
INDIKATIONEN
SUSTIVA® (Efavirenz) in Kombination mit anderen antiretroviralen Wirkstoffen ist indiziert zur Behandlung einer Infektion mit dem humanen Immunschwächevirus Typ 1 (HIV-1) bei Erwachsenen und pädiatrischen Patienten ab einem Alter von 3 Monaten und einem Körpergewicht von mindestens 3,5 kg.
DOSIERUNG UND ANWENDUNG
Leberfunktion
Überwachen Sie die Leberfunktion vor und während der Behandlung mit SUSTIVA [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. SUSTIVA 600 mg wird bei Patienten mit mäßiger oder schwerer Leberfunktionsstörung (Child Pugh B oder C) nicht empfohlen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Erwachsene
Die empfohlene Dosierung von SUSTIVA (Efavirenz) beträgt 600 mg oral einmal täglich in Kombination mit einem Proteasehemmer und/oder nukleosidanalogen Reverse-Transkriptase-Hemmern (NRTIs). Es wird empfohlen, SUSTIVA auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Die nach Einnahme von SUSTIVA 600 mg mit Nahrung beobachteten erhöhten Efavirenz-Konzentrationen können zu einer Zunahme der Häufigkeit von Nebenwirkungen führen [siehe KLINISCHE PHARMAKOLOGIE ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit von Symptomen des Nervensystems verbessern [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , NEBENWIRKUNGEN , und INFORMATIONEN ZUM PATIENTEN ]. SUSTIVA Kapseln oder Tabletten sollten unzerkaut mit Flüssigkeit geschluckt werden. Für Patienten, die keine Kapseln oder Tabletten schlucken können, wird die Verabreichungsmethode mit Kapselspritzern empfohlen [siehe Kapsel besprühen Art der Verabreichung ].
Begleitende antiretrovirale Therapie
SUSTIVA muss in Kombination mit anderen antiretroviralen Medikamenten gegeben werden [siehe INDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN , und KLINISCHE PHARMAKOLOGIE ].
Dosisanpassung
Wenn SUSTIVA 200 mg zusammen mit Voriconazol angewendet wird, sollte die Voriconazol-Erhaltungsdosis auf 400 mg alle 12 Stunden erhöht und die SUSTIVA 200 mg-Dosis auf 300 mg einmal täglich unter Verwendung der Kapselformulierung (eine 200-mg- und zwei 50-mg-Kapseln oder sechs 50-mg-Kapseln) verringert werden mg-Kapseln). SUSTIVA 600 mg Tabletten dürfen nicht zerbrochen werden. [sehen WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE .]
Wenn SUSTIVA zusammen mit Rifampin an Patienten verabreicht wird, die 50 kg oder mehr wiegen, wird eine Erhöhung der Dosis von SUSTIVA 200 mg auf 800 mg einmal täglich empfohlen [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ].
Pädiatrische Patienten
Es wird empfohlen, SUSTIVA 600 mg auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Tabelle 1 beschreibt die empfohlene Dosis von SUSTIVA für pädiatrische Patienten im Alter von 3 Monaten oder älter und einem Gewicht zwischen 3,5 kg und 40 kg [siehe KLINISCHE PHARMAKOLOGIE ]. Die empfohlene Dosierung von SUSTIVA für pädiatrische Patienten mit einem Körpergewicht von 40 kg oder mehr beträgt 600 mg einmal täglich. Für pädiatrische Patienten, die keine Kapseln schlucken können, kann der Kapselinhalt mit einer kleinen Menge Nahrung oder Säuglingsanfangsnahrung unter Anwendung der Kapsel-Sprinkle-Methode verabreicht werden [siehe Kapsel besprühen Art der Verabreichung ].
Kapsel besprühen Art der Verabreichung
Bei pädiatrischen Patienten ab einem Alter von 3 Monaten und einem Gewicht von mindestens 3,5 kg sowie bei Erwachsenen, die Kapseln oder Tabletten nicht schlucken können, kann der Kapselinhalt mit einer kleinen Menge (1 bis 2 Teelöffel) Nahrung eingenommen werden. Die Verwendung von Säuglingsanfangsnahrung zum Mischen sollte nur für junge Säuglinge in Betracht gezogen werden, die feste Nahrung nicht zuverlässig zu sich nehmen können. Patienten und Pflegepersonal sollten angewiesen werden, die Kapsel vorsichtig zu öffnen, um ein Verschütten oder Verteilen des Kapselinhalts in die Luft zu vermeiden. Die Kapsel sollte waagerecht über einen kleinen Behälter gehalten und zum Öffnen vorsichtig gedreht werden. Bei Patienten, die feste Nahrung vertragen, sollte der gesamte Kapselinhalt in dem kleinen Behälter vorsichtig mit einer altersgerechten weichen Nahrung wie Apfelmus, Traubengelee oder Joghurt vermischt werden. Bei kleinen Säuglingen, die die Kapsel-Streusel-Säuglingsnahrung-Mischung erhalten, sollte der gesamte Kapselinhalt vorsichtig in 2 Teelöffel rekonstituierte Säuglingsnahrung bei Raumtemperatur in einem kleinen Behälter gemischt werden, indem vorsichtig mit einem kleinen Löffel umgerührt und die Mischung dann in eine 10 ml orale Dosierspritze zur Verabreichung. Nach der Verabreichung der SUSTIVA-Nahrungs- oder Nahrungsmischung muss eine zusätzliche kleine Menge (ca. 2 Teelöffel) Nahrung oder Nahrungsnahrung in den leeren Mischbehälter gegeben, gerührt werden, um alle verbleibenden SUSTIVA-Reste aufzulösen, und dem Patienten verabreicht werden. Die SUSTIVA-Nahrungs- oder Formelmischung sollte innerhalb von 30 Minuten nach dem Mischen verabreicht werden. Für 2 Stunden nach der Verabreichung von SUSTIVA sollte keine zusätzliche Nahrung aufgenommen werden.
Weitere Patientenhinweise zur Verabreichungsmethode mit Kapselstreuung finden sich in der von der FDA zugelassenen Patientenkennzeichnung (siehe INFORMATIONEN ZUM PATIENTEN und GEBRAUCHSANWEISUNG ).
WIE GELIEFERT
Darreichungsformen und Stärken
Kapseln
Die 200-mg-Kapseln sind goldfarben, mit dem Rückseitenaufdruck „SUSTIVA“ auf dem Unterteil und dem Aufdruck „200 mg“ auf dem Oberteil.
Die 50-mg-Kapseln sind goldfarben und weiß, mit dem Aufdruck „SUSTIVA“ auf dem goldfarbenen Oberteil und dem umgekehrten Aufdruck „50 mg“ auf dem weißen Unterteil.
Tablets
600-mg-Tabletten sind gelbe, kapselförmige Filmtabletten mit dem Aufdruck „SUSTIVA“ auf beiden Seiten.
Lagerung und Handhabung
Kapseln
SUSTIVA® (Efavirenz) Kapseln sind wie folgt erhältlich:
Die 200-mg-Kapseln sind goldfarben, mit dem Rückseitenaufdruck „SUSTIVA“ auf dem Unterteil und dem Aufdruck „200 mg“ auf dem Oberteil.
Flaschen von 90 - NDC 0056-0474-92
Die 50-mg-Kapseln sind goldfarben und weiß, mit dem Aufdruck „SUSTIVA“ auf dem goldfarbenen Oberteil und dem umgekehrten Aufdruck „50 mg“ auf dem weißen Unterteil.
Flaschen von 30 - NDC 0056-0470-30
Tablets
SUSTIVA® (Efavirenz) Tabletten sind wie folgt erhältlich:
Tabletten 600 mg sind gelbe, kapselförmige Filmtabletten mit dem Aufdruck „SUSTIVA“ auf beiden Seiten.
Flaschen von 30 - NDC 0056-0510-30
Lagerung
SUSTIVA 600 mg Kapseln und SUSTIVA Tabletten sollten bei 25 °C (77 °F) gelagert werden; Abweichungen bis 15°C-30°C (59°F-86°F) zulässig [siehe USP Controlled Room Temperature].
Vertrieb durch: Bristol-Myers Squibb Company Princeton, NJ 08543 USA. Überarbeitet: Okt. 2020
NEBENWIRKUNGEN
Die wichtigsten Nebenwirkungen, die bei mit SUSTIVA behandelten Patienten beobachtet wurden, sind:
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die berichteten Nebenwirkungsraten nicht direkt mit den Raten in anderen klinischen Studien verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Nebenwirkungen bei Erwachsenen
Die häufigsten (> 5 % in beiden Efavirenz-Behandlungsgruppen) Nebenwirkungen von mindestens mäßigem Schweregrad bei Patienten in Studie 006, die mit SUSTIVA 200 mg in Kombination mit Zidovudin/Lamivudin oder Indinavir behandelt wurden, waren Hautausschlag, Schwindel, Übelkeit, Kopfschmerzen, Müdigkeit, Schlaflosigkeit, und Erbrechen.
Ausgewählte klinische Nebenwirkungen mittlerer oder schwerer Intensität, die bei ≥ 2 % der mit SUSTIVA behandelten Patienten in zwei kontrollierten klinischen Studien beobachtet wurden, sind in Tabelle 2 aufgeführt.
Pankreatitis wurde berichtet, obwohl ein kausaler Zusammenhang mit Efavirenz nicht nachgewiesen wurde. Asymptomatische Anstiege der Amylasespiegel im Serum wurden bei einer signifikant höheren Anzahl von mit Efavirenz 600 mg behandelten Patienten beobachtet als bei Kontrollpatienten (vgl Laboranomalien ).
Symptome des Nervensystems
Für 1008 Patienten, die mit SUSTIVA-haltigen Regimen behandelt wurden, und 635 Patienten, die in kontrollierten Studien mit einem Kontrollregime behandelt wurden, listet Tabelle 3 die Häufigkeit von Symptomen unterschiedlicher Schweregrade auf und gibt die Abbruchraten für eines oder mehrere der folgenden Symptome des Nervensystems an: Schwindel, Schlaflosigkeit, Konzentrationsstörungen, Somnolenz, anormales Träumen, Euphorie, Verwirrtheit, Erregung, Amnesie, Halluzinationen, Benommenheit, anormales Denken und Depersonalisation [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Häufigkeiten spezifischer Symptome des zentralen und peripheren Nervensystems sind in Tabelle 2 angegeben.
Psychiatrische Symptome
Bei mit SUSTIVA behandelten Patienten wurde über schwerwiegende psychiatrische Nebenwirkungen berichtet. In kontrollierten Studien waren Depression (19 %, 16 %), Angst (13 %, 9 %) und Nervosität (7 %) psychiatrische Symptome, die mit einer Häufigkeit von mehr als 2 % bei mit SUSTIVA 600 mg bzw. Kontrollregimen behandelten Patienten beobachtet wurden. , 2 %).
Ausschlag
In kontrollierten klinischen Studien betrug die Häufigkeit von Hautausschlägen (alle Schweregrade, unabhängig von der Kausalität) 26 % bei 1008 Erwachsenen, die mit Therapien behandelt wurden, die SUSTIVA 200 mg enthielten, und 17 % bei 635 Erwachsenen, die mit einer Kontrolltherapie behandelt wurden. Die meisten Berichte über Hautausschlag waren leicht oder mittelschwer. Die Häufigkeit von Hautausschlag Grad 3 betrug 0,8 % bei mit SUSTIVA behandelten Patienten und 0,3 % bei den Kontrollgruppen, und die Häufigkeit von Hautausschlag Grad 4 betrug 0,1 % bei SUSTIVA und 0 bei Kontrollgruppen. Die Abbruchraten aufgrund von Hautausschlag betrugen 1,7 % bei den mit SUSTIVA behandelten Patienten und 0,3 % bei den Kontrollgruppen [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Die Erfahrungen mit SUSTIVA bei Patienten, die andere antiretrovirale Arzneimittel der NNRTI-Klasse abgesetzt haben, sind begrenzt. Neunzehn Patienten, die Nevirapin wegen Hautausschlag abgesetzt hatten, wurden mit SUSTIVA behandelt. Neun dieser Patienten entwickelten während der Therapie mit SUSTIVA einen leichten bis mittelschweren Hautausschlag, und zwei dieser Patienten brachen die Behandlung wegen Hautausschlag ab.
Laboranomalien
Ausgewählte Laboranomalien der Grade 3-4, die bei ≥ 2 % der mit SUSTIVA behandelten Patienten in zwei klinischen Studien berichtet wurden, sind in Tabelle 4 aufgeführt.
Patienten, die gleichzeitig mit Hepatitis B oder C infiziert sind
Leberfunktionstests sollten bei Patienten mit Hepatitis B und/oder C in der Anamnese überwacht werden. Im Langzeitdatensatz von Studie 006 wurden 137 Patienten mit SUSTIVA-haltigen Regimen behandelt (mittlere Therapiedauer 68 Wochen) und 84 behandelt mit einem Kontrollregime (mittlere Dauer 56 Wochen) waren beim Screening auf Hepatitis B (Oberflächenantigen-positiv) und/oder C (Hepatitis-C-Antikörper-positiv) seropositiv. Unter diesen koinfizierten Patienten traten bei 13 % der Patienten in den SUSTIVA-Armen und 7 % der Patienten im Kontrollarm Erhöhungen der AST auf mehr als das Fünffache der ULN auf, und bei 20 % der Patienten traten Erhöhungen der ALT auf mehr als das Fünffache der ULN auf in den SUSTIVA-Armen und 7 % der Patienten im Kontrollarm. Unter den koinfizierten Patienten brachen 3 % der mit SUSTIVA-haltigen Regimen behandelten und 2 % im Kontrollarm die Studie wegen Leber- oder Gallenwegserkrankungen ab [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Lipide
Bei einigen nicht infizierten Probanden, die SUSTIVA erhielten, wurde ein Anstieg des Gesamtcholesterins gegenüber dem Ausgangswert um 10-20 % beobachtet. Bei Patienten, die mit SUSTIVA + Zidovudin + Lamivudin behandelt wurden, wurden Anstiege von ungefähr 20 % bzw. 25 % gegenüber dem Ausgangswert von Gesamtcholesterin und HDL ohne Nüchternblut beobachtet. Bei Patienten, die mit SUSTIVA + Indinavir behandelt wurden, wurden Anstiege von Cholesterin und HDL außerhalb des Nüchternzustands um etwa 40 % bzw. 35 % gegenüber dem Ausgangswert beobachtet. Gesamtcholesterinspiegel außerhalb des Nüchternzustands von ≥ 240 mg/dl und ≥ 300 mg/dl wurden bei 34 % bzw. 9 % der mit SUSTIVA + Zidovudin + Lamivudin behandelten Patienten berichtet; 54 % bzw. 20 % der mit SUSTIVA + Indinavir behandelten Patienten; und 28 % bzw. 4 % der mit Indinavir + Zidovudin + Lamivudin behandelten Patienten. Die Wirkungen von SUSTIVA 600 mg auf Triglyceride und LDL in dieser Studie waren nicht gut charakterisiert, da die Proben von nicht nüchternen Patienten genommen wurden. Die klinische Bedeutung dieser Befunde ist unbekannt [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Nebenwirkungen bei pädiatrischen Patienten
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die berichteten Nebenwirkungsraten nicht direkt mit den Raten in anderen klinischen Studien verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Die Bewertung der Nebenwirkungen basiert auf drei klinischen Studien mit 182 HIV-1-infizierten pädiatrischen Patienten (im Alter von 3 Monaten bis 21 Jahren), die SUSTIVA 200 mg in Kombination mit anderen antiretroviralen Wirkstoffen über einen medianen Zeitraum von 123 Wochen erhielten. Die in den drei Studien beobachteten Nebenwirkungen ähnelten denen, die in klinischen Studien bei Erwachsenen beobachtet wurden, mit der Ausnahme, dass Hautausschlag häufiger bei pädiatrischen Patienten auftrat (32 % für alle Schweregrade, unabhängig von der Kausalität) und häufiger von höherem Schweregrad (dh schwerer). Bei zwei (1,1 %) pädiatrischen Patienten kam es zu einem Hautausschlag Grad 3 (konfluierender Hautausschlag mit Fieber, generalisierter Hautausschlag) und bei vier (2,2 %) pädiatrischen Patienten kam es zu einem Hautausschlag Grad 4 (alles Erythema multiforme). Fünf pädiatrische Patienten (2,7 %) brachen die Studie wegen Hautausschlag ab [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von SUSTIVA nach der Zulassung festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet wurden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Körper als Ganzes: allergische Reaktionen, Asthenie, Umverteilung/Ansammlung von Körperfett [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]
Zentrales und peripheres Nervensystem: Koordinationsstörungen, Ataxie, Enzephalopathie, zerebelläre Koordinations- und Gleichgewichtsstörungen, Krämpfe, Hypästhesie, Parästhesien, Neuropathie, Tremor, Schwindel
Endokrin: Gynäkomastie
Magen-Darm: Verstopfung, Malabsorption
Herz-Kreislauf: Erröten, Herzklopfen
Leber und Gallensystem: Anstieg der Leberenzyme, Leberversagen, Hepatitis.
Stoffwechsel und Ernährung: Hypercholesterinämie, Hypertriglyzeridämie
Bewegungsapparat: Arthralgie, Myalgie, Myopathie
Psychiatrie: aggressive Reaktionen, Erregung, Wahnvorstellungen, emotionale Labilität, Manie, Neurose, Paranoia, Psychose, Selbstmord, Katatonie
Atmung: Dyspnoe
Haut und Anhängsel: Erythema multiforme, photoallergische Dermatitis, Stevens-Johnson-Syndrom
Besondere Sinne: Sehstörungen, Tinnitus
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Mögliche Auswirkungen von SUSTIVA auf andere Medikamente
Es wurde gezeigt, dass Efavirenz in vivo CYP3A und CYP2B6 induziert. Andere Verbindungen, die Substrate von CYP3A oder CYP2B6 sind, können verringerte Plasmakonzentrationen aufweisen, wenn sie zusammen mit SUSTIVA verabreicht werden.
Mögliche Beeinflussung von SUSTIVA durch andere Medikamente
Es ist zu erwarten, dass Arzneimittel, die eine CYP3A-Aktivität induzieren (z. B. Phenobarbital, Rifampin, Rifabutin), die Clearance von Efavirenz erhöhen, was zu verringerten Plasmakonzentrationen führt [siehe DOSIERUNG UND ANWENDUNG ].
QT-verlängernde Medikamente
Es liegen nur begrenzte Informationen über die Möglichkeit einer pharmakodynamischen Wechselwirkung zwischen SUSTIVA 600 mg und Arzneimitteln vor, die das QTc-Intervall verlängern. Bei Anwendung von Efavirenz wurde eine QTc-Verlängerung beobachtet [siehe KLINISCHE PHARMAKOLOGIE ]. Ziehen Sie Alternativen zu SUSTIVA 600 mg in Betracht, wenn es zusammen mit einem Medikament verabreicht wird, bei dem ein bekanntes Torsade-de-Pointes-Risiko besteht.
Gegründete und andere potenziell signifikante Wechselwirkungen mit anderen Arzneimitteln
Arzneimittelwechselwirkungen mit SUSTIVA sind in Tabelle 5 zusammengefasst. Für Daten zur Pharmakokinetik [siehe KLINISCHE PHARMAKOLOGIE ] Tabellen 7 und 8. Diese Tabelle enthält potenziell signifikante Wechselwirkungen, ist jedoch nicht vollständig.
Arzneimittel ohne klinisch signifikante Wechselwirkungen mit SUSTIVA
Es wird keine Dosisanpassung empfohlen, wenn SUSTIVA 600 mg zusammen mit den folgenden Arzneimitteln gegeben wird: Aluminium-/Magnesiumhydroxid-Antazida, Azithromycin, Cetirizin, Famotidin, Fluconazol, Lorazepam, Nelfinavir, nukleosidische Reverse-Transkriptase-Hemmer (Abacavir, Emtricitabin, Lamivudin, Stavudin, Tenofovirdisoproxilfumarat, Zidovudin). ), Paroxetin und Raltegravir.
Cannabinoid-Test-Interaktion
Efavirenz bindet nicht an Cannabinoidrezeptoren. Bei einigen Screening-Assays bei nicht infizierten und HIV-infizierten Probanden, die Efavirenz erhielten, wurde über falsch-positive Cannabinoid-Testergebnisse im Urin berichtet. Die Bestätigung positiver Screening-Tests für Cannabinoide durch eine spezifischere Methode wird empfohlen.
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Wechselwirkungen mit anderen Medikamenten
Die Plasmakonzentrationen von Efavirenz können durch Substrate, Inhibitoren oder Induktoren von CYP3A verändert werden. Ebenso kann Efavirenz die Plasmakonzentrationen von Arzneimitteln verändern, die durch CYP3A oder CYP2B6 metabolisiert werden. Die hervorstechendste Wirkung von Efavirenz im Steady State ist die Induktion von CYP3A und CYP2B6 [vgl DOSIERUNG UND ANWENDUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
QTc-Verlängerung
Bei Anwendung von Efavirenz wurde eine QTc-Verlängerung beobachtet [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ]. Ziehen Sie Alternativen zu SUSTIVA 600 mg in Betracht, wenn es zusammen mit einem Arzneimittel verabreicht wird, bei dem ein bekanntes Risiko für Torsade de Pointes besteht, oder wenn es Patienten mit einem höheren Risiko für Torsade de Pointes verabreicht wird.
Widerstand
SUSTIVA 200 mg darf nicht als Einzelwirkstoff zur Behandlung einer HIV-1-Infektion angewendet oder als Einzelwirkstoff zu einem versagenden Regime hinzugefügt werden. Resistente Viren treten schnell auf, wenn Efavirenz als Monotherapie verabreicht wird. Bei der Auswahl neuer antiretroviraler Wirkstoffe, die in Kombination mit Efavirenz angewendet werden sollen, sollte das Potenzial für eine virale Kreuzresistenz berücksichtigt werden.
Koadministration mit verwandten Produkten
Die gleichzeitige Anwendung von SUSTIVA mit ATRIPLA (Efavirenz 600 mg/Emtricitabin 200 mg/Tenofovirdisoproxilfumarat 300 mg) wird nicht empfohlen, es sei denn, dies ist zur Dosisanpassung erforderlich (z. B. mit Rifampin), da Efavirenz einer seiner Wirkstoffe ist.
Psychiatrische Symptome
Bei mit SUSTIVA behandelten Patienten wurde über schwerwiegende psychiatrische Nebenwirkungen berichtet. In kontrollierten Studien mit 1008 Patienten, die durchschnittlich 2,1 Jahre lang mit 600 mg SUSTIVA-Regimen behandelt wurden, und 635 Patienten, die durchschnittlich 1,5 Jahre lang mit Kontrollschemata behandelt wurden, wurde die Häufigkeit (unabhängig von der Kausalität) spezifischer schwerwiegender psychiatrischer Ereignisse bei Patienten, die SUSTIVA erhielten, oder Kontrollschemata waren schwere Depressionen (2,4 %, 0,9 %), Selbstmordgedanken (0,7 %, 0,3 %), nicht tödliche Selbstmordversuche (0,5 %, 0), aggressives Verhalten (0,4 %, 0,5 %), paranoide Reaktionen (0,4 %, 0,3 %) und manische Reaktionen (0,2 %, 0,3 %). Wenn ähnliche psychiatrische Symptome wie die oben genannten kombiniert und in einer multifaktoriellen Analyse der Daten aus Studie 006 als Gruppe ausgewertet wurden, war die Behandlung mit Efavirenz mit einem vermehrten Auftreten dieser ausgewählten psychiatrischen Symptome verbunden. Andere Faktoren, die mit einem Anstieg des Auftretens dieser psychiatrischen Symptome verbunden sind, waren die Vorgeschichte des intravenösen Drogenkonsums, die psychiatrische Vorgeschichte und die Einnahme von psychiatrischen Medikamenten bei Studieneintritt; ähnliche Assoziationen wurden sowohl in der SUSTIVA- als auch in der Kontrollgruppe beobachtet. In Studie 006 traten während der gesamten Studie sowohl bei den mit SUSTIVA behandelten als auch bei den Kontrollpatienten neue schwerwiegende psychiatrische Symptome auf. Ein Prozent der mit SUSTIVA behandelten Patienten hat die Behandlung wegen eines oder mehrerer dieser ausgewählten psychiatrischen Symptome abgebrochen oder unterbrochen. Es gab auch gelegentliche Postmarketing-Berichte über Tod durch Suizid, Wahnvorstellungen und psychoseähnliches Verhalten, obwohl aus diesen Berichten kein kausaler Zusammenhang mit der Anwendung von SUSTIVA ermittelt werden kann. Postmarketing-Fälle von Katatonie wurden ebenfalls berichtet und können mit einer erhöhten Efavirenz-Exposition in Zusammenhang stehen. Patienten mit schwerwiegenden psychiatrischen Nebenwirkungen sollten sich umgehend medizinisch untersuchen lassen, um die Möglichkeit abzuschätzen, dass die Symptome mit der Anwendung von SUSTIVA in Zusammenhang stehen könnten, und falls ja, um festzustellen, ob die Risiken einer fortgesetzten Therapie den Nutzen überwiegen. [sehen NEBENWIRKUNGEN .]
Symptome des Nervensystems
Dreiundfünfzig Prozent (531/1008) der Patienten, die SUSTIVA 600 mg in kontrollierten Studien erhielten, berichteten über Symptome des Zentralnervensystems (jeden Grades, unabhängig von der Kausalität), verglichen mit 25 % (156/635) der Patienten, die Kontrollregime erhielten [siehe NEBENWIRKUNGEN ]. Zu diesen Symptomen gehörten unter anderem Schwindel (28,1 % der 1008 Patienten), Schlaflosigkeit (16,3 %), Konzentrationsstörungen (8,3 %), Somnolenz (7,0 %), abnormale Träume (6,2 %) und Halluzinationen (1.2 %). Diese Symptome waren bei 2,0 % der Patienten schwerwiegend; und 2,1 % der Patienten brachen daraufhin die Therapie ab. Diese Symptome beginnen normalerweise am ersten oder zweiten Therapietag und klingen im Allgemeinen nach den ersten 2-4 Wochen der Therapie ab. Nach 4-wöchiger Therapie lag die Prävalenz von Symptomen des Nervensystems von mindestens mäßigem Schweregrad zwischen 5 % und 9 % bei Patienten, die mit SUSTIVA-haltigen Regimen behandelt wurden, und zwischen 3 % und 5 % bei Patienten, die mit einem Kontrollregime behandelt wurden. Die Patienten sollten darüber informiert werden, dass sich diese häufigen Symptome bei Fortführung der Therapie wahrscheinlich bessern und nicht vorhersagend für das spätere Auftreten der weniger häufigen psychiatrischen Symptome sind [siehe Psychiatrische Symptome ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit dieser Symptome des Nervensystems verbessern [siehe DOSIERUNG UND ANWENDUNG ].
Die Analyse der Langzeitdaten aus Studie 006 (mediane Nachbeobachtungszeit 180 Wochen, 102 Wochen bzw. 76 Wochen für Patienten, die mit SUSTIVA + Zidovudin + Lamivudin, SUSTIVA + Indinavir bzw. Indinavir + Zidovudin + Lamivudin behandelt wurden) zeigte dies darüber hinaus Nach 24-wöchiger Therapie war die Inzidenz neu aufgetretener Symptome des Nervensystems bei den mit SUSTIVA behandelten Patienten im Allgemeinen ähnlich wie im Indinavir-enthaltenden Kontrollarm.
Eine spät einsetzende Neurotoxizität, einschließlich Ataxie und Enzephalopathie (Bewusstseinsstörungen, Verwirrtheit, psychomotorische Verlangsamung, Psychose, Delirium), kann Monate bis Jahre nach Beginn der Efavirenz-Therapie auftreten. Bei Patienten mit genetischen CYP2B6-Polymorphismen, die trotz Standarddosierung von SUSTIVA mit erhöhten Efavirenz-Spiegeln einhergehen, traten einige Ereignisse mit spät einsetzender Neurotoxizität auf. Patienten mit Anzeichen und Symptomen schwerwiegender neurologischer Nebenwirkungen sollten umgehend untersucht werden, um die Möglichkeit zu beurteilen, dass diese Ereignisse mit der Anwendung von Efavirenz in Zusammenhang stehen und ob ein Absetzen von SUSTIVA 600 mg gerechtfertigt ist.
Patienten, die SUSTIVA 600 mg erhalten, sollten auf die Möglichkeit zusätzlicher Wirkungen auf das Zentralnervensystem aufmerksam gemacht werden, wenn SUSTIVA 200 mg gleichzeitig mit Alkohol oder Psychopharmaka angewendet wird.
Patienten mit zentralnervösen Symptomen wie Schwindel, Konzentrationsstörungen und/oder Schläfrigkeit sollten potenziell gefährliche Tätigkeiten wie Autofahren oder Bedienen von Maschinen vermeiden.
Embryo-fötale Toxizität
Efavirenz kann den Fötus schädigen, wenn es einer schwangeren Frau während des ersten Trimenons verabreicht wird. Weisen Sie gebärfähige Frauen, die SUSTIVA 600 mg erhalten, darauf hin, eine Schwangerschaft zu vermeiden. [sehen Verwendung in bestimmten Bevölkerungsgruppen .]
Ausschlag
In kontrollierten klinischen Studien traten bei 26 % (266/1008) der erwachsenen Patienten, die mit 600 mg SUSTIVA behandelt wurden, neue Hautausschläge auf, verglichen mit 17 % (111/635) der in den Kontrollgruppen behandelten Patienten [siehe NEBENWIRKUNGEN ]. Bei 0,9 % (9/1008) der mit SUSTIVA behandelten Patienten trat ein mit Blasenbildung, feuchter Abschuppung oder Ulzeration einhergehender Hautausschlag auf. Die Inzidenz von Ausschlag 4. Grades (z. B. Erythema multiforme, Stevens-Johnson-Syndrom) bei erwachsenen Patienten, die mit SUSTIVA in allen Studien und erweitertem Zugang behandelt wurden, betrug 0,1 %. Hautausschläge sind in der Regel leichte bis mittelschwere makulopapulöse Hautausschläge, die innerhalb der ersten 2 Wochen nach Beginn der Therapie mit Efavirenz auftreten (die mediane Zeit bis zum Auftreten des Hautausschlags bei Erwachsenen betrug 11 Tage) und bei den meisten Patienten, die die Therapie mit Efavirenz fortsetzen, verschwindet der Hautausschlag innerhalb von 1 Tag Monat (mittlere Dauer 16 Tage). Die Abbruchrate wegen Hautausschlag in klinischen Studien mit Erwachsenen betrug 1,7 % (17/1008).
Ausschlag wurde bei 59 von 182 pädiatrischen Patienten (32 %), die mit SUSTIVA behandelt wurden, berichtet [siehe NEBENWIRKUNGEN ]. Bei zwei pädiatrischen Patienten kam es zu einem Hautausschlag Grad 3 (konfluierender Hautausschlag mit Fieber, generalisierter Hautausschlag), und vier Patienten hatten einen Hautausschlag Grad 4 (Erythema multiforme). Die mediane Zeit bis zum Auftreten des Hautausschlags bei pädiatrischen Patienten betrug 28 Tage (Bereich 3–1642 Tage). Vor Beginn der Therapie mit SUSTIVA bei pädiatrischen Patienten sollte eine Prophylaxe mit geeigneten Antihistaminika in Betracht gezogen werden.
SUSTIVA 600 mg kann im Allgemeinen bei Patienten wieder aufgenommen werden, die die Therapie aufgrund eines Hautausschlags unterbrechen. SUSTIVA 200 mg sollte bei Patienten abgesetzt werden, die einen schweren Hautausschlag entwickeln, der mit Blasenbildung, Abschuppung, Schleimhautbeteiligung oder Fieber einhergeht. Geeignete Antihistaminika und/oder Kortikosteroide können die Verträglichkeit verbessern und das Abklingen des Hautausschlags beschleunigen. Bei Patienten, die eine lebensbedrohliche Hautreaktion (z. B. Stevens-Johnson-Syndrom) hatten, sollte eine alternative Therapie in Betracht gezogen werden [siehe KONTRAINDIKATIONEN ].
Hepatotoxizität
Postmarketing-Fälle von Hepatitis, einschließlich fulminanter Hepatitis, die zu einem Leberversagen fortschreiten, das eine Transplantation erfordert oder zum Tod führt, wurden bei mit SUSTIVA behandelten Patienten berichtet. Die Berichte umfassten Patienten mit zugrunde liegender Lebererkrankung, einschließlich Koinfektion mit Hepatitis B oder C, und Patienten ohne vorbestehende Lebererkrankung oder andere identifizierbare Risikofaktoren.
SUSTIVA 600 mg wird für Patienten mit mäßiger oder schwerer Leberfunktionsstörung nicht empfohlen. Bei Patienten mit leichter Leberfunktionsstörung, die SUSTIVA erhalten, wird eine sorgfältige Überwachung empfohlen. [sehen NEBENWIRKUNGEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Eine Überwachung der Leberenzyme vor und während der Behandlung wird für alle Patienten empfohlen [siehe DOSIERUNG UND ANWENDUNG ].
Erwägen Sie das Absetzen von SUSTIVA 600 mg bei Patienten mit anhaltenden Erhöhungen der Serumtransaminasen auf mehr als das Fünffache der Obergrenze des Normalbereichs.Setzen Sie SUSTIVA 200 mg ab, wenn die Erhöhung der Serumtransaminasen von klinischen Anzeichen oder Symptomen einer Hepatitis oder Leberdekompensation begleitet wird.
Krämpfe
Bei erwachsenen und pädiatrischen Patienten, die Efavirenz erhielten, wurden Konvulsionen beobachtet, im Allgemeinen bei Vorliegen bekannter Anfälle in der Krankengeschichte [siehe Nichtklinische Toxikologie ]. Vorsicht ist geboten bei allen Patienten mit Krampfanfällen in der Anamnese. Bei Patienten, die gleichzeitig Antikonvulsiva erhalten, die hauptsächlich von der Leber metabolisiert werden, wie Phenytoin und Phenobarbital, kann eine regelmäßige Überwachung der Plasmaspiegel erforderlich sein [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Lipiderhöhungen
Die Behandlung mit SUSTIVA hat zu einem Anstieg der Konzentration von Gesamtcholesterin und Triglyceriden geführt [siehe NEBENWIRKUNGEN ]. Cholesterin- und Triglyceridtests sollten vor Beginn der Therapie mit SUSTIVA 200 mg und in regelmäßigen Abständen während der Therapie durchgeführt werden.
Immunrekonstitutionssyndrom
Bei Patienten, die mit einer antiretroviralen Kombinationstherapie, einschließlich SUSTIVA, behandelt wurden, wurde über ein Immunrekonstitutionssyndrom berichtet. Während der Anfangsphase einer antiretroviralen Kombinationsbehandlung können Patienten, deren Immunsystem anspricht, eine entzündliche Reaktion auf indolente oder verbleibende opportunistische Infektionen (wie Mycobacterium avium-Infektion, Cytomegalovirus, Pneumocystis-jiroveci-Pneumonie [PCP] oder Tuberkulose) entwickeln, die eine weitere Untersuchung erforderlich machen kann und Behandlung.
Es wurde auch über das Auftreten von Autoimmunerkrankungen (wie Morbus Basedow, Polymyositis, Guillain-Barré-Syndrom und Autoimmunhepatitis) im Zusammenhang mit der Immunrekonstitution berichtet; Die Zeit bis zum Einsetzen ist jedoch variabler und kann viele Monate nach Beginn der Behandlung auftreten.
Fettumverteilung
Bei Patienten, die eine antiretrovirale Therapie erhielten, wurde eine Umverteilung/Ansammlung von Körperfett einschließlich zentraler Adipositas, dorsozervikaler Fettvergrößerung (Büffelbuckel), peripherer Auszehrung, Gesichtsauszehrung, Brustvergrößerung und „cushingoidem Aussehen“ beobachtet. Der Mechanismus und die langfristigen Folgen dieser Ereignisse sind derzeit unbekannt. Ein kausaler Zusammenhang wurde nicht hergestellt.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( INFORMATIONEN ZUM PATIENTEN und Gebrauchsanweisung ).
Wechselwirkungen mit anderen Medikamenten
Auf den Flaschenetiketten des Produkts befindet sich eine Erklärung für Patienten und medizinisches Fachpersonal: ACHTUNG: Informieren Sie sich über Arzneimittel, die NICHT zusammen mit SUSTIVA eingenommen werden sollten.
SUSTIVA kann mit einigen Arzneimitteln interagieren; raten Sie den Patienten daher, die Verwendung anderer verschreibungspflichtiger oder nicht verschreibungspflichtiger Medikamente ihrem Arzt zu melden.
Allgemeine Informationen für Patienten
Informieren Sie die Patienten darüber, dass SUSTIVA 200 mg keine Heilung für eine HIV-1-Infektion darstellt und die Patienten möglicherweise weiterhin an Krankheiten im Zusammenhang mit einer HIV-1-Infektion leiden, einschließlich opportunistischer Infektionen. Die Patienten sollten während der Einnahme von SUSTIVA in ärztlicher Behandlung bleiben.
Raten Sie den Patienten, Dinge zu vermeiden, die eine HIV-1-Infektion auf andere übertragen können.
Dosierungsanleitung
Empfehlen Sie den Patienten, SUSTIVA jeden Tag wie verordnet einzunehmen. Wenn ein Patient die Einnahme von SUSTIVA vergisst, sagen Sie ihm, dass er die vergessene Dosis sofort einnehmen soll, es sei denn, es ist fast Zeit für die nächste Dosis. Weisen Sie den Patienten darauf hin, nicht 2 Dosen auf einmal einzunehmen und die nächste Dosis zum regulär geplanten Zeitpunkt einzunehmen. Raten Sie dem Patienten, einen Arzt zu fragen, ob er/sie Hilfe bei der Planung der besten Zeiten für die Einnahme seiner/ihrer Medikamente benötigt.
SUSTIVA 200 mg muss immer in Kombination mit anderen antiretroviralen Arzneimitteln angewendet werden. Empfehlen Sie den Patienten, SUSTIVA 600 mg auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Die Einnahme von SUSTIVA zusammen mit Nahrung erhöht die Konzentration von Efavirenz und kann die Häufigkeit von Nebenwirkungen erhöhen. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit von Symptomen des Nervensystems verbessern [siehe DOSIERUNG UND ANWENDUNG und NEBENWIRKUNGEN ]. Gesundheitsdienstleister sollten Eltern oder Betreuungspersonen bei der Bestimmung des besten SUSTIVA-Dosierungsplans für Säuglinge und Kleinkinder unterstützen.
Für erwachsene und pädiatrische Patienten, die keine Kapseln oder Tabletten schlucken können, sollten Patienten oder ihre Betreuungspersonen angewiesen werden, die Anweisungen zur Verabreichung des Kapselinhalts in einer kleinen Menge Nahrung oder Säuglingsanfangsnahrung zu lesen und sorgfältig zu befolgen [siehe DOSIERUNG UND ANWENDUNG und FDA-zugelassene Patientenkennzeichnung ( INFORMATIONEN ZUM PATIENTEN und GEBRAUCHSANWEISUNG )]. Patienten sollten ihren Arzt oder Apotheker anrufen, wenn sie Fragen haben.
Symptome des Nervensystems
Informieren Sie die Patienten darüber, dass Symptome des Zentralnervensystems (NSS), einschließlich Schwindel, Schlaflosigkeit, Konzentrationsstörungen, Benommenheit und abnormale Träume, häufig während der ersten Wochen der Therapie mit SUSTIVA berichtet werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit dieser Symptome verbessern, die sich wahrscheinlich bei fortgesetzter Therapie bessern werden. Machen Sie die Patienten auf mögliche additive Wirkungen aufmerksam, wenn SUSTIVA gleichzeitig mit Alkohol oder Psychopharmaka angewendet wird. Weisen Sie die Patienten darauf hin, dass sie potenziell gefährliche Tätigkeiten wie Autofahren oder Bedienen von Maschinen vermeiden sollten, wenn sie NSS erleben.
Informieren Sie die Patienten darüber, dass ein Risiko für die Entwicklung einer spät einsetzenden Neurotoxizität besteht, einschließlich Ataxie und Enzephalopathie, die Monate bis Jahre nach Beginn der SUSTIVA-Therapie auftreten können [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Psychiatrische Symptome
Informieren Sie die Patienten darüber, dass bei Patienten, die SUSTIVA erhielten, über schwerwiegende psychiatrische Symptome einschließlich schwerer Depression, Suizidversuche, aggressives Verhalten, Wahnvorstellungen, Paranoia, psychoseähnliche Symptome und Katatonie berichtet wurde [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Wenn sie schwerwiegende psychiatrische Nebenwirkungen erfahren, sollten sie sich unverzüglich medizinisch untersuchen lassen. Raten Sie den Patienten, ihren Arzt über jede Vorgeschichte von psychischen Erkrankungen oder Drogenmissbrauch zu informieren.
Ausschlag
Informieren Sie die Patienten darüber, dass Hautausschlag eine häufige Nebenwirkung ist [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Hautausschläge verschwinden in der Regel ohne Änderung der Behandlung. Da der Hautausschlag jedoch schwerwiegend sein kann, raten Sie den Patienten, sich unverzüglich an ihren Arzt zu wenden, wenn ein Hautausschlag auftritt.
Hepatotoxizität
Weisen Sie die Patienten darauf hin, auf frühe Warnzeichen einer Leberentzündung oder eines Leberversagens, wie Müdigkeit, Schwäche, Appetitlosigkeit, Übelkeit und Erbrechen, sowie auf spätere Anzeichen wie Gelbsucht, Verwirrtheit, Bauchschwellung und verfärbten Kot zu achten und ihren Arzt zu konsultieren unverzüglich einen Arzt aufsuchen, wenn solche Symptome auftreten [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Weibchen mit reproduktivem Potenzial
Raten Sie gebärfähigen Frauen, während der Behandlung mit SUSTIVA 600 mg und für 12 Wochen nach Absetzen von SUSTIVA eine wirksame Verhütungsmethode sowie eine Barrieremethode anzuwenden. Weisen Sie Patientinnen darauf hin, sich an ihren Arzt zu wenden, wenn sie eine Schwangerschaft planen, schwanger werden oder wenn während der Behandlung mit SUSTIVA eine Schwangerschaft vermutet wird [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Schwangerschafts-Expositionsregister
Weisen Sie die Patientinnen darauf hin, dass es ein Schwangerschaftsexpositionsregister gibt, das die Schwangerschaftsausgänge bei Frauen überwacht, die während der Schwangerschaft SUSTIVA ausgesetzt waren [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Fettumverteilung
Informieren Sie die Patienten darüber, dass es bei Patienten, die eine antiretrovirale Therapie erhalten, zu einer Umverteilung oder Ansammlung von Körperfett kommen kann und dass die Ursache und die langfristigen gesundheitlichen Auswirkungen dieser Erkrankungen nicht bekannt sind [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
SUSTIVA ist eine eingetragene Marke der Bristol-Myers Squibb Pharma Company. ATRIPLA ist eine Marke von Bristol-Myers Squibb & Gilead Sciences, LLC.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Mit Efavirenz wurden Langzeitstudien zur Kanzerogenität an Mäusen und Ratten durchgeführt. Mäuse wurden 2 Jahre lang mit 0, 25, 75, 150 oder 300 mg/kg/Tag behandelt. Die Inzidenzen von hepatozellulären Adenomen und Karzinomen sowie pulmonalen alveolären/bronchiolären Adenomen waren bei Frauen über den Normalwert hinaus erhöht. Bei Männern wurde kein Anstieg der Tumorinzidenz über den Hintergrund hinaus beobachtet. Für diese Studie wurde kein NOAEL bei Frauen festgelegt, da bei allen Dosierungen Tumorbefunde auftraten. Die AUC beim NOAEL (150 mg/kg) war bei Männern etwa 0,9-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis. In der Studie an Ratten wurde bei Dosen bis zu 100 mg/kg/Tag, bei denen die AUCs das 0,1- (Männer) oder 0,2-Fache (Frauen) der empfohlenen klinischen Dosis betrugen, kein Anstieg der Tumorinzidenz beobachtet.
Mutagenese
Efavirenz wurde in einer Reihe von In-vitro- und In-vivo-Genotoxizitätstests negativ getestet. Diese umfassten bakterielle Mutationsassays in S. typhimurium und E. coli, Mutationsassays von Säugetieren in Eierstockzellen des chinesischen Hamsters, Chromosomenaberrationsassays in humanen peripheren Blutlymphozyten oder Eierstockzellen des chinesischen Hamsters und ein In-vivo-Maus-Knochenmark-Mikronukleus-Assay.
Beeinträchtigung der Fruchtbarkeit
Efavirenz beeinträchtigte weder die Paarung noch die Fertilität männlicher oder weiblicher Ratten und beeinträchtigte nicht die Spermien behandelter männlicher Ratten. Die Fortpflanzungsfähigkeit von Nachkommen weiblicher Ratten, denen Efavirenz verabreicht wurde, war nicht beeinträchtigt. Die AUCs bei den NOAEL-Werten bei männlichen (200 mg/kg) und weiblichen (100 mg/kg) Ratten waren etwa ≤0,15-mal höher als beim Menschen bei der empfohlenen klinischen Dosis.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschafts-Expositionsregister
Es gibt ein Schwangerschafts-Expositionsregister, das den Ausgang der Schwangerschaft bei Frauen überwacht, die während der Schwangerschaft SUSTIVA 600 mg ausgesetzt waren. Ärzte werden ermutigt, Patienten anzumelden, indem sie das Antiretroviral Pregnancy Registry unter 1-800-258-4263 anrufen.
Zusammenfassung der Risiken
Es gibt retrospektive Fallberichte über Neuralrohrdefekte bei Säuglingen, deren Mütter im ersten Trimenon der Schwangerschaft Efavirenz-haltigen Therapien ausgesetzt waren. Prospektive Schwangerschaftsdaten aus dem Antiretroviral Pregnancy Registry reichen nicht aus, um dieses Risiko angemessen einzuschätzen. Verfügbare Daten aus dem Antiretroviral Pregnancy Registry zeigen keinen Unterschied im Gesamtrisiko für schwere Geburtsfehler im Vergleich zur Hintergrundrate für schwere Geburtsfehler von 2,7 % in der US-Referenzpopulation des Metropolitan Atlanta Congenital Defects Program (MACDP). Obwohl kein kausaler Zusammenhang zwischen der Efavirenz-Exposition im ersten Trimenon und Neuralrohrdefekten hergestellt werden konnte, wurden ähnliche Missbildungen in Studien beobachtet, die an Affen mit ähnlichen Dosen wie beim Menschen durchgeführt wurden. Darüber hinaus traten bei Ratten bei einer Dosis, die zehnmal geringer war als die menschliche Exposition bei der empfohlenen klinischen Dosis, fötale und embryonale Toxizitäten auf. Aufgrund des potenziellen Risikos von Neuralrohrdefekten sollte Efavirenz im ersten Trimenon der Schwangerschaft nicht angewendet werden. Informieren Sie schwangere Frauen über das potenzielle Risiko für einen Fötus.
Daten
Menschliche Daten
Es gibt retrospektive Postmarketing-Berichte über Befunde, die mit Neuralrohrdefekten übereinstimmen, einschließlich Meningomyelozele, alle bei Säuglingen von Müttern, die im ersten Trimenon Efavirenz-haltigen Behandlungen ausgesetzt waren.
Basierend auf prospektiven Berichten des Antiretroviral Pregnancy Registry (APR) von etwa 1000 Lebendgeburten nach Exposition gegenüber Efavirenz-haltigen Regimen (einschließlich über 800 Lebendgeburten, die im ersten Trimenon exponiert wurden), gab es keinen Unterschied zwischen Efavirenz und Geburtsfehlern insgesamt im Vergleich zu Efavirenz Hintergrund-Geburtsfehlerrate von 2,7 % in der US-Referenzpopulation des Metropolitan Atlanta Congenital Defects Program. Gemäß dem im Dezember 2014 herausgegebenen Zwischenbericht zum effektiven Jahreszins lag die Prävalenz von Geburtsfehlern nach einer Exposition im ersten Trimester bei 2,3 % (95 %-KI: 1,4 %–3,6 %). Einer dieser prospektiv berichteten Defekte bei Exposition im ersten Trimester war ein Neuralrohrdefekt. Ein einzelner Fall von Anophthalmie mit Efavirenz-Exposition im ersten Trimester wurde ebenfalls prospektiv berichtet. Dieser Fall umfasste auch schwere schräge Gesichtsspalten und Fruchtwasserstreifen, die bekanntermaßen mit Anophthalmie in Verbindung stehen.
Tierdaten
Die Auswirkungen von Efavirenz auf die embryofetale Entwicklung wurden bei drei nichtklinischen Spezies (Cynomolgus-Affen, Ratten und Kaninchen) untersucht. Bei Affen wurde Efavirenz 60 mg/kg/Tag trächtigen Weibchen während der gesamten Trächtigkeit (20. bis 150. Trächtigkeitstag) verabreicht. Die maternale systemische Arzneimittelexposition (AUC) betrug das 1,3-fache der Exposition beim Menschen bei der empfohlenen klinischen Dosis (600 mg/Tag), wobei die fötalen Nabelvenen-Arzneimittelkonzentrationen etwa das 0,7-fache der maternalen Werte betrugen. Drei von 20 Föten/Säuglingen hatten eine oder mehrere Missbildungen; Es gab keine missgebildeten Föten oder Säuglinge von mit Placebo behandelten Müttern. Die Missbildungen, die bei diesen drei Affenföten auftraten, umfassten Anenzephalie und einseitige Anophthalmie bei einem Fötus, Mikrophthalmie bei einem zweiten und eine Gaumenspalte bei dem dritten. Für diese Studie wurde kein NOAEL (No Observable Adverse Effect Level) festgelegt, da nur eine Dosierung bewertet wurde. Bei Ratten wurde Efavirenz entweder während der Organogenese (7. bis 18. Trächtigkeitstag) oder vom 7. Trächtigkeitstag bis zum 21. Laktationstag mit 50, 100 oder 200 mg/kg/Tag verabreicht. Die Verabreichung von 200 mg/kg/Tag an Ratten war mit einem Anstieg der Inzidenz früher Resorptionen verbunden; und Dosen von 100 mg/kg/Tag und mehr waren mit früher neonataler Sterblichkeit verbunden. Die AUC beim NOAEL (50 mg/kg/Tag) war in dieser Rattenstudie 0,1-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis. Die Wirkstoffkonzentrationen in der Milch am 10. Laktationstag waren etwa 8-mal höher als die im mütterlichen Plasma. Bei trächtigen Kaninchen war Efavirenz weder embryoletal noch teratogen, wenn es in Dosierungen von 25, 50 und 75 mg/kg/Tag während der Organogenese (6. bis 18. Trächtigkeitstag) verabreicht wurde. Die AUC beim NOAEL (75 mg/kg/Tag) bei Kaninchen war 0,4-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis.
Stillzeit
Zusammenfassung der Risiken
Die Centers for Disease Control and Prevention empfehlen HIV-infizierten Müttern, ihre Säuglinge nicht zu stillen, um eine postnatale Übertragung von HIV zu vermeiden. Wegen der Möglichkeit einer HIV-Übertragung bei gestillten Säuglingen raten Sie Frauen vom Stillen ab.
Weibchen und Männchen mit reproduktivem Potenzial
Wegen möglicher teratogener Wirkungen sollte eine Schwangerschaft bei Frauen, die SUSTIVA erhalten, vermieden werden. [sehen Schwangerschaft .]
Schwangerschaftstest
Frauen im gebärfähigen Alter sollten sich vor Beginn der Behandlung mit SUSTIVA einem Schwangerschaftstest unterziehen.
Empfängnisverhütung
Frauen im gebärfähigen Alter sollten aufgrund der langen Halbwertszeit von Efavirenz während der Behandlung mit SUSTIVA 200 mg und für 12 Wochen nach Absetzen von SUSTIVA 200 mg eine wirksame Verhütungsmethode anwenden. Die Barriere-Kontrazeption sollte immer in Kombination mit anderen Verhütungsmethoden angewendet werden. Hormonelle Methoden, die Progesteron enthalten, können eine verminderte Wirksamkeit haben [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Pädiatrische Verwendung
Die Sicherheit, das pharmakokinetische Profil sowie das virologische und immunologische Ansprechen von SUSTIVA 600 mg wurden bei antiretroviral-naiven und -erfahrenen HIV-1-infizierten pädiatrischen Patienten im Alter von 3 Monaten bis 21 Jahren in drei offenen klinischen Studien untersucht [siehe NEBENWIRKUNGEN , KLINISCHE PHARMAKOLOGIE , und Klinische Studien ]. Die Art und Häufigkeit von Nebenwirkungen in diesen Studien war im Allgemeinen ähnlich wie bei erwachsenen Patienten, mit Ausnahme einer höheren Häufigkeit von Hautausschlag, einschließlich einer höheren Häufigkeit von Hautausschlag Grad 3 oder 4, bei pädiatrischen Patienten im Vergleich zu Erwachsenen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Die Anwendung von SUSTIVA bei Patienten unter 3 Monaten ODER unter 3,5 kg Körpergewicht wird nicht empfohlen, da die Sicherheit, Pharmakokinetik und antivirale Aktivität von SUSTIVA 600 mg in dieser Altersgruppe nicht untersucht wurden und das Risiko besteht, eine HIV-Resistenz zu entwickeln wenn SUSTIVA unterdosiert wird. sehen DOSIERUNG UND ANWENDUNG für Dosierungsempfehlungen für pädiatrische Patienten.
Geriatrische Verwendung
Klinische Studien mit SUSTIVA 200 mg schlossen keine ausreichende Anzahl von Probanden ab 65 Jahren ein, um festzustellen, ob sie anders reagieren als jüngere Probanden. Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein, um die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und von Begleiterkrankungen oder anderen Therapien widerzuspiegeln.
Leberfunktionsstörung
SUSTIVA wird für Patienten mit mäßiger oder schwerer Leberfunktionsstörung nicht empfohlen, da keine ausreichenden Daten vorliegen, um festzustellen, ob eine Dosisanpassung erforderlich ist. Patienten mit leichter Leberfunktionsstörung können ohne Dosisanpassung mit Efavirenz behandelt werden. Aufgrund des umfangreichen Cytochrom-P450-vermittelten Metabolismus von Efavirenz und begrenzter klinischer Erfahrung bei Patienten mit eingeschränkter Leberfunktion ist bei der Verabreichung von SUSTIVA an diese Patienten Vorsicht geboten [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Einige Patienten, die versehentlich zweimal täglich 600 mg eingenommen haben, haben über verstärkte Symptome des Nervensystems berichtet. Bei einem Patienten traten unwillkürliche Muskelkontraktionen auf.
Die Behandlung einer Überdosierung mit SUSTIVA 200 mg sollte aus allgemeinen unterstützenden Maßnahmen bestehen, einschließlich Überwachung der Vitalfunktionen und Beobachtung des klinischen Zustands des Patienten. Die Verabreichung von Aktivkohle kann verwendet werden, um die Entfernung von nicht resorbiertem Arzneimittel zu unterstützen. Es gibt kein spezifisches Antidot für eine Überdosierung mit SUSTIVA. Da Efavirenz stark an Proteine gebunden ist, ist es unwahrscheinlich, dass eine Dialyse das Arzneimittel signifikant aus dem Blut entfernt.
KONTRAINDIKATIONEN
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Efavirenz ist ein antivirales Medikament [siehe Mikrobiologie ].
Pharmakodynamik
Herzelektrophysiologie
Die Wirkung von SUSTIVA 600 mg auf das QTc-Intervall wurde in einer unverblindeten, positiven und Placebo-kontrollierten Crossover-QT-Studie mit fester Einzelsequenz über 3 Perioden und 3 Behandlungen bei 58 gesunden Probanden, die mit CYP2B6-Polymorphismen angereichert waren, untersucht. Die mittlere Cmax von Efavirenz bei Patienten mit dem CYP2B6 *6/*6-Genotyp nach Verabreichung einer Tagesdosis von 600 mg über 14 Tage war 2,25-mal höher als die mittlere Cmax, die bei Patienten mit dem CYP2B6 *1/*1-Genotyp beobachtet wurde. Es wurde eine positive Beziehung zwischen der Efavirenz-Konzentration und der QTc-Verlängerung beobachtet. Basierend auf der Konzentrations-QTc-Beziehung betragen die mittlere QTc-Verlängerung und ihr oberes 90 %-Konfidenzintervall 8,7 ms und 11,3 ms bei Patienten mit CYP2B6*6/*6-Genotyp nach Verabreichung einer Tagesdosis von 600 mg über 14 Tage [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Pharmakokinetik
Absorption
Maximale Efavirenz-Plasmakonzentrationen von 1,6 – 9,1 μM wurden 5 Stunden nach Verabreichung einer oralen Einzeldosis von 100 mg bis 1600 mg an nicht infizierte Probanden erreicht. Dosisabhängige Anstiege von Cmax und AUC wurden bei Dosen bis zu 1600 mg beobachtet; die Anstiege waren weniger als proportional, was auf eine verringerte Absorption bei höheren Dosen hindeutet.
Bei HIV-1-infizierten Patienten im Steady State waren die mittlere Cmax, mittlere Cmin und mittlere AUC nach Tagesdosen von 200 mg, 400 mg und 600 mg dosisproportional. Die Zeit bis zum Erreichen der maximalen Plasmakonzentrationen betrug etwa 3-5 Stunden und Steady-State-Plasmakonzentrationen wurden in 6-10 Tagen erreicht. Bei 35 Patienten, die SUSTIVA 600 mg einmal täglich erhielten, betrug die Steady-State-Cmax 12,9 ± 3,7 μM (Mittelwert ± Standardabweichung), die Steady-State-Cmin 5,6 ± 3,2 μM und die AUC 184 ± 73 μM•h.
Einfluss der Nahrung auf die orale Aufnahme:
Kapseln
Die Einnahme einer Einzeldosis von 600 mg Efavirenz-Kapseln mit einer fettreichen/kalorienreichen Mahlzeit (894 kcal, 54 g Fett, 54 % Kalorien aus Fett) oder einer fettreduzierten/normalkalorischen Mahlzeit (440 kcal, 2 g Fett, 4 % Kalorien aus Fett) war mit einem mittleren Anstieg der AUC∞ von Efavirenz um 22 % bzw. 17 % und einem mittleren Anstieg der Cmax von Efavirenz um 39 % bzw. 51 % verbunden, relativ zu den Expositionen, die bei Verabreichung im nüchternen Zustand erreicht wurden . [sehen DOSIERUNG UND ANWENDUNG und INFORMATIONEN ZUM PATIENTEN .]
Tablets
Die Einnahme einer einzelnen 600-mg-Efavirenz-Tablette mit einer fettreichen/kalorienreichen Mahlzeit (ca. 1000 kcal, 500-600 kcal aus Fett) war mit einem 28 %igen Anstieg der mittleren AUC∞ von Efavirenz und einem 79 %igen Anstieg des Mittelwertes verbunden Cmax von Efavirenz im Verhältnis zu den unter nüchternen Bedingungen erzielten Expositionen. [sehen DOSIERUNG UND ANWENDUNG und INFORMATIONEN ZUM PATIENTEN .]
Bioverfügbarkeit von Kapselinhalten gemischt mit Nahrungsvehikeln
Bei gesunden erwachsenen Probanden erfüllte die AUC von Efavirenz bei Verabreichung als Inhalt von drei 200-mg-Kapseln gemischt mit 2 Teelöffeln bestimmter Nahrungsvehikel (Apfelmus, Traubengelee oder Joghurt oder Säuglingsnahrung) die Bioäquivalenzkriterien für die AUC der verabreichten intakten Kapselformulierung unter nüchternen Bedingungen.
Verteilung
Efavirenz wird in hohem Maße (ca. 99,5-99,75 %) an menschliche Plasmaproteine, überwiegend Albumin, gebunden. Bei HIV-1-infizierten Patienten (n = 9), die SUSTIVA 200 bis 600 mg einmal täglich für mindestens einen Monat erhielten, lagen die Konzentrationen im Liquor cerebrospinalis im Bereich von 0,26 bis 1,19 % (Mittelwert 0,69 %) der entsprechenden Plasmakonzentration. Dieser Anteil ist etwa 3-mal höher als der nicht proteingebundene (freie) Anteil von Efavirenz im Plasma.
Stoffwechsel
Studien am Menschen und In-vitro-Studien mit menschlichen Lebermikrosomen haben gezeigt, dass Efavirenz hauptsächlich durch das Cytochrom-P450-System zu hydroxylierten Metaboliten mit anschließender Glucuronidierung dieser hydroxylierten Metaboliten metabolisiert wird. Diese Metaboliten sind gegenüber HIV-1 im Wesentlichen inaktiv. Die In-vitro-Studien deuten darauf hin, dass CYP3A und CYP2B6 die wichtigsten Isozyme sind, die für den Metabolismus von Efavirenz verantwortlich sind.
Es wurde gezeigt, dass Efavirenz CYP-Enzyme induziert, was zu einer Induktion seines eigenen Metabolismus führt. Mehrfachdosen von 200–400 mg pro Tag über 10 Tage führten zu einem geringeren Ausmaß der Akkumulation als erwartet (22–42 % niedriger) und einer kürzeren terminalen Halbwertszeit von 40–55 Stunden (Halbwertszeit einer Einzeldosis 52–76 Stunden). ).
Beseitigung
Efavirenz hat eine terminale Halbwertszeit von 52-76 Stunden nach Einzeldosen und 40-55 Stunden nach Mehrfachdosen. Es wurde eine einmonatige Massenbilanz/Ausscheidungsstudie mit 400 mg pro Tag mit einer C-markierten Dosis durchgeführt, die an Tag 8 verabreicht wurde. Ungefähr 14-34 % der radioaktiv markierten Substanz wurden im Urin und 16-61 % im Stuhl wiedergefunden . Fast die gesamte Urinausscheidung des radioaktiv markierten Arzneimittels erfolgte in Form von Metaboliten. Efavirenz machte den größten Teil der gesamten im Stuhl gemessenen Radioaktivität aus.
Besondere Populationen
Pädiatrie
Die pharmakokinetischen Parameter für Efavirenz im Steady State bei pädiatrischen Patienten wurden durch ein populationspharmakokinetisches Modell vorhergesagt und sind in Tabelle 6 nach Gewichtsbereichen zusammengefasst, die den empfohlenen Dosen entsprechen.
Geschlecht und Rasse
Die Pharmakokinetik von Efavirenz bei Patienten scheint bei Männern und Frauen sowie bei den untersuchten ethnischen Gruppen ähnlich zu sein.
Nierenfunktionsstörung
Die Pharmakokinetik von Efavirenz wurde bei Patienten mit Niereninsuffizienz nicht untersucht; jedoch wird weniger als 1 % von Efavirenz unverändert im Urin ausgeschieden, sodass die Auswirkungen einer Nierenfunktionsstörung auf die Efavirenz-Elimination minimal sein sollten.
Leberfunktionsstörung
Eine Mehrfachdosisstudie zeigte keine signifikante Wirkung auf die Pharmakokinetik von Efavirenz bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) im Vergleich zu Kontrollen. Es lagen keine ausreichenden Daten vor, um festzustellen, ob eine mäßige oder schwere Leberfunktionsstörung (Child-Pugh-Klasse B oder C) die Pharmakokinetik von Efavirenz beeinflusst.
Arzneimittelwechselwirkungsstudien
Es wurde in vivo gezeigt, dass Efavirenz eine hepatische Enzyminduktion verursacht und somit die Biotransformation einiger Arzneimittel erhöht, die durch CYP3A und CYP2B6 metabolisiert werden. In-vitro-Studien haben gezeigt, dass Efavirenz die CYP-Isoenzyme 2C9 und 2C19 mit Ki-Werten (8,5-17 μM) im Bereich der beobachteten Efavirenz-Plasmakonzentrationen hemmt. In In-vitro-Studien hemmte Efavirenz CYP2E1 nicht und hemmte CYP2D6 und CYP1A2 (Ki-Werte 82-160 μM) nur bei Konzentrationen, die deutlich über den klinisch erreichten liegen. Die gleichzeitige Anwendung von Efavirenz mit Arzneimitteln, die hauptsächlich durch CYP2C9-, CYP2C19-, CYP3A- oder CYP2B6-Isozyme metabolisiert werden, kann zu veränderten Plasmakonzentrationen des gleichzeitig verabreichten Arzneimittels führen. Es ist zu erwarten, dass Arzneimittel, die CYP3A- und CYP2B6-Aktivität induzieren, die Clearance von Efavirenz erhöhen, was zu verringerten Plasmakonzentrationen führt.
Wechselwirkungsstudien wurden mit Efavirenz und anderen Arzneimitteln durchgeführt, die wahrscheinlich gleichzeitig verabreicht werden, oder Arzneimitteln, die üblicherweise als Sonden für pharmakokinetische Wechselwirkungen verwendet werden. Die Wirkungen der gleichzeitigen Verabreichung von Efavirenz auf Cmax, AUC und Cmin sind in Tabelle 7 (Wirkung von Efavirenz auf andere Arzneimittel) und Tabelle 8 (Wirkung anderer Arzneimittel auf Efavirenz) zusammengefasst. Informationen zu klinischen Empfehlungen finden Sie unter WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN .
Mikrobiologie
Wirkmechanismus
Efavirenz ist ein NNRTI von HIV-1. Die Aktivität von Efavirenz wird überwiegend durch nicht-kompetitive Hemmung der Reversen Transkriptase von HIV-1 vermittelt. Die reverse Transkriptase von HIV-2 und die humanen zellulären DNA-Polymerasen α, β, γ und δ werden durch Efavirenz nicht gehemmt.
Antivirale Aktivität in der Zellkultur
Die Konzentration von Efavirenz, die die Replikation von laboradaptierten Wildtypstämmen und klinischen Isolaten in Zellkulturen um 90-95 % (EC90-95) hemmt, lag im Bereich von 1,7 bis 25 nM in lymphoblastoiden Zelllinien, peripheren mononukleären Blutzellen (PBMCs) und Makrophagen /Monozytenkulturen. Efavirenz zeigte eine antivirale Aktivität gegen Klade B- und die meisten Nicht-Klade B-Isolate (Subtypen A, AE, AG, C, D, F, G, J, N), hatte aber eine reduzierte antivirale Aktivität gegen Gruppe-O-Viren. Efavirenz zeigte in Zellkultur eine additive antivirale Aktivität ohne Zytotoxizität gegen HIV-1, wenn es mit den NNRTIs Delavirdin und Nevirapin, NRTIs (Abacavir, Didanosin, Emtricitabin, Lamivudin, Stavudin, Tenofovir, Zalcitabin, Zidovudin), PIs (Amprenavir, Indinavir, Lopinavir, Nelfinavir, Ritonavir, Saquinavir) und der Fusionsinhibitor Enfuvirtid. Efavirenz zeigte in Zellkultur mit Atazanavir eine additive bis antagonistische antivirale Aktivität. Efavirenz war nicht antagonistisch mit Adefovir, das zur Behandlung einer Hepatitis-B-Virusinfektion angewendet wird, oder mit Ribavirin, das in Kombination mit Interferon zur Behandlung einer Hepatitis-C-Virusinfektion angewendet wird.
Widerstand
In Zellkultur
In Zellkulturen traten in Gegenwart des Arzneimittels schnell HIV-1-Isolate mit reduzierter Empfindlichkeit gegenüber Efavirenz (>380-facher Anstieg des EC90-Werts) auf. Die genotypische Charakterisierung dieser Viren identifizierte einzelne Aminosäuresubstitutionen L100I oder V179D, doppelte Substitutionen L100I/V108I und dreifache Substitutionen L100I/V179D/Y181C in der reversen Transkriptase.
Klinische Studien
Klinische Isolate mit reduzierter Empfindlichkeit gegenüber Efavirenz in Zellkulturen wurden erhalten. Eine oder mehrere Substitutionen an den Aminosäurepositionen 98, 100, 101, 103, 106, 108, 188, 190, 225 und 227 in der Reversen Transkriptase wurden bei Patienten beobachtet, bei denen die Behandlung mit Efavirenz in Kombination mit Indinavir oder mit Zidovudin plus Lamivudin fehlschlug. Die K103N-Substitution wurde am häufigsten beobachtet. In der langfristigen Resistenzüberwachung (durchschnittlich 52 Wochen, Bereich 4–106 Wochen) wurden 28 Isolate mit übereinstimmenden Ausgangswerten und virologischem Versagen analysiert. Einundsechzig Prozent (17/28) dieser fehlgeschlagenen Isolate hatten eine verringerte Efavirenz-Empfindlichkeit in der Zellkultur mit einer medianen 88-fachen Veränderung der Efavirenz-Empfindlichkeit (EC50-Wert) gegenüber der Referenz. Die häufigste NNRTI-Substitution, die sich bei diesen Patientenisolaten entwickelte, war K103N (54 %). Andere NNRTI-Substitutionen, die sich entwickelten, waren L100I (7 %), K101E/Q/R (14 %), V108I (11 %), G190S/T/A (7 %), P225H (18 %) und M230I/L (11 %).
Kreuzresistenz
Kreuzresistenz zwischen NNRTIs wurde beobachtet. Klinische Isolate, die zuvor als Efavirenz-resistent charakterisiert wurden, waren im Vergleich zum Ausgangswert in Zellkulturen auch phänotypisch resistent gegen Delavirdin und Nevirapin. Delavirdin- und/oder Nevirapin-resistente klinische Virusisolate mit NNRTI-Resistenz-assoziierten Substitutionen (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L oder M230L) zeigten eine reduzierte Empfindlichkeit gegenüber Efavirenz in Zellkultur. Mehr als 90 % der in Zellkultur getesteten NRTI-resistenten klinischen Isolate behielten ihre Empfindlichkeit gegenüber Efavirenz.
Tiertoxikologie
Nicht anhaltende Krämpfe wurden bei 6 von 20 Affen beobachtet, die Efavirenz in Dosen erhielten, die Plasma-AUC-Werte ergaben, die 4- bis 13-mal höher waren als beim Menschen, der die empfohlene Dosis erhielt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Klinische Studien
Erwachsene
Studie 006 eine randomisierte, offene Studie, verglich SUSTIVA (600 mg einmal täglich) + Zidovudin (ZDV, 300 mg alle 12 Stunden) + Lamivudin (LAM, 150 mg alle 12 Stunden) oder SUSTIVA (600 mg einmal täglich) + Indinavir (IDV, 1000 mg q8h) mit Indinavir (800 mg q8h) + Zidovudin (300 mg q12h) + Lamivudin (150 mg q12h). Zwölfhundertsechsundsechzig Patienten (Durchschnittsalter 36,5 Jahre [Bereich 18–81], 60 % Kaukasier, 83 % männlich) wurden eingeschlossen. Alle Patienten waren bei Studieneintritt Efavirenz-, Lamivudin-, NNRTI- und PI-naiv. Die mediane CD4+-Zellzahl zu Studienbeginn betrug 320 Zellen/mm3 und die mediane HIV-1-RNA-Konzentration zu Studienbeginn betrug 4,8 log Kopien/ml. Die Behandlungsergebnisse mit dem Standard-Assay (Assay-Grenze 400 Kopien/ml) über 48 und 168 Wochen sind in Tabelle 9 dargestellt. Versionen des AMPLICOR HIV-1 MONITOR Assays. Während der Studie wurde Version 1.5 des Assays in Europa eingeführt, um den Nachweis von Nicht-Clade-B-Viren zu verbessern.
Bei Patienten, die mit SUSTIVA + Zidovudin + Lamivudin, SUSTIVA + Indinavir oder Indinavir + Zidovudin + Lamivudin behandelt wurden, betrug der Prozentsatz der Responder mit HIV-1-RNA
ACTG 364 ist eine randomisierte, doppelblinde, placebokontrollierte 48-wöchige Studie an NRTI-erfahrenen Patienten, die zwei vorherige ACTG-Studien abgeschlossen hatten. Einhundertsechsundneunzig Patienten (Durchschnittsalter 41 Jahre [Bereich 18–76], 74 % Kaukasier, 88 % männlich) erhielten NRTIs in Kombination mit SUSTIVA (600 mg einmal täglich) oder Nelfinavir (NFV, 750 mg dreimal täglich). ) oder SUSTIVA (600 mg einmal täglich) + Nelfinavir randomisiert und doppelblind verabreicht. Die mittlere CD4+-Zellzahl zu Studienbeginn betrug 389 Zellen/mm3 und die mittlere HIV-1-RNA-Konzentration zu Studienbeginn 8130 Kopien/ml. Bei Eintritt in die Studie wurde allen Patienten ein neues offenes NRTI-Regime zugewiesen, das von ihrer bisherigen NRTI-Behandlungserfahrung abhängig war. Es gab keinen signifikanten Unterschied in der mittleren CD4+-Zellzahl zwischen den Behandlungsgruppen; die durchschnittliche Gesamtzunahme betrug etwa 100 Zellen nach 48 Wochen bei Patienten, die die Studienbehandlungen fortsetzten. Die Behandlungsergebnisse sind in Tabelle 10 aufgeführt. Die Plasma-HIV-RNA-Spiegel wurden mit dem AMPLICOR HIV-1 MONITOR-Assay unter Verwendung einer unteren Quantifizierungsgrenze von 500 Kopien/ml quantifiziert.
Eine Kaplan-Meier-Analyse der Zeit bis zum Therapieversagen über 72 Wochen zeigt eine längere Dauer der virologischen Suppression (HIV-RNA
Pädiatrische Patienten
Studie AI266922 ist eine Open-Label-Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA 200 mg in Kombination mit Didanosin und Emtricitabin bei antiretroviral-naiven und -erfahrenen pädiatrischen Patienten. 37 Patienten im Alter von 3 Monaten bis 6 Jahren (Median 0,7 Jahre) wurden mit SUSTIVA behandelt. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 5,88 log Kopien/ml, die mediane CD4+-Zellzahl 1144 Zellen/mm3 und der mediane CD4+-Prozentsatz 25 %. Die mediane Dauer der Studientherapie betrug 60 Wochen; 27 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrugen die Gesamtanteile von Patienten mit HIV-RNA
Studie PACTG 1021 war eine offene Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA 200 mg in Kombination mit Didanosin und Emtricitabin bei pädiatrischen Patienten, die keine antiretrovirale Therapie erhalten hatten. 43 Patienten im Alter von 3 Monaten bis 21 Jahren (Median 9,6 Jahre) erhielten eine Dosis von SUSTIVA. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 4,8 log Kopien/ml, die mediane CD4+-Zellzahl 367 Zellen/mm3 und der mediane CD4+-Prozentsatz 18 %. Die mediane Dauer der Studientherapie betrug 181 Wochen; 16 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrugen die Gesamtanteile von Patienten mit HIV-RNA
Studie PACTG 382 war eine offene Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA in Kombination mit Nelfinavir und einem NRTI bei antiretroviral-naiven und NRTI-erfahrenen pädiatrischen Patienten. 102 Patienten im Alter von 3 Monaten bis 16 Jahren (Median 5,7 Jahre) wurden mit SUSTIVA behandelt. 87 % der Patienten hatten zuvor eine antiretrovirale Therapie erhalten. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 4,57 log Kopien/ml, die mediane CD4+-Zellzahl 755 Zellen/mm3 und der mediane CD4+-Prozentsatz 30 %. Die mediane Dauer der Studientherapie betrug 118 Wochen; 25 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrug der Gesamtanteil der Patienten mit HIV-RNA
INFORMATIONEN ZUM PATIENTEN
SUSTIVA® (sus-TEE-vah) (Efavirenz) Kapseln
SUSTIVA® (sus-TEE-vah) (Efavirenz) Tabletten
Wichtig: Fragen Sie Ihren Arzt oder Apotheker nach Arzneimitteln, die Sie nicht zusammen mit SUSTIVA einnehmen sollten. Weitere Informationen finden Sie im Abschnitt „Was soll ich meinem Arzt vor der Einnahme von SUSTIVA mitteilen?“
Lesen Sie diese Patienteninformation, bevor Sie mit der Einnahme von SUSTIVA beginnen und jedes Mal, wenn Sie eine Nachfüllung erhalten. Möglicherweise gibt es neue Informationen. Diese Informationen ersetzen nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung.
Was ist SUSTIVA?
SUSTIVA 600 mg ist ein verschreibungspflichtiges HIV-1-Arzneimittel (Humanes Immunschwächevirus Typ 1), das zusammen mit anderen antiretroviralen Arzneimitteln zur Behandlung einer HIV-1-Infektion bei Erwachsenen und Kindern angewendet wird, die mindestens 3 Monate alt sind und mindestens 7 Pfund 12 Unzen (3,5 kg) wiegen kg). HIV ist das Virus, das AIDS (Acquired Immune Deficiency Syndrome) verursacht.
Es ist nicht bekannt, ob SUSTIVA bei Kindern unter 3 Monaten oder mit einem Gewicht von weniger als 3,5 kg (7 Pfund 12 Unzen) sicher und wirksam ist.
Bei Anwendung mit anderen antiretroviralen Arzneimitteln zur Behandlung einer HIV-1-Infektion kann SUSTIVA 600 mg helfen:
Die Reduzierung der HIV-1-Menge und die Erhöhung der CD4+ (T)-Zellen in Ihrem Blut kann zur Verbesserung Ihres Immunsystems beitragen. Dies kann Ihr Risiko zu sterben oder Infektionen zu bekommen, die auftreten können, wenn Ihr Immunsystem geschwächt ist (opportunistische Infektionen).
SUSTIVA 600 mg heilt weder eine HIV-1-Infektion noch AIDS. Sie sollten weiterhin HIV-1-Medikamente einnehmen, um eine HIV-1-Infektion zu kontrollieren und HIV-bedingte Erkrankungen zu verringern.
Vermeiden Sie Dinge, die eine HIV-1-Infektion auf andere übertragen können:
Fragen Sie Ihren Arzt, wenn Sie Fragen dazu haben, wie Sie verhindern können, dass HIV auf andere Menschen übertragen wird.
Wer sollte SUSTIVA 600 mg nicht einnehmen?
Nehmen Sie SUSTIVA nicht ein, wenn Sie allergisch gegen Efavirenz oder einen der sonstigen Bestandteile von SUSTIVA sind. Eine vollständige Liste der Inhaltsstoffe von SUSTIVA finden Sie am Ende dieser Packungsbeilage.
Nehmen Sie SUSTIVA nicht ein, wenn Sie derzeit Elbasvir und Grazoprevir (ZEPATIER®) einnehmen.
Was sollte ich meinem Arzt vor der Einnahme von SUSTIVA 200 mg mitteilen?
Informieren Sie vor der Einnahme von SUSTIVA Ihren Arzt, wenn Sie an einer Erkrankung leiden und insbesondere, wenn Sie:
Informieren Sie Ihren Arzt und Apotheker über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und Kräuterergänzungen.
SUSTIVA 600 mg kann die Wirkungsweise anderer Arzneimittel beeinflussen, und andere Arzneimittel können die Wirkungsweise von SUSTIVA 600 mg beeinflussen. und kann schwerwiegende Nebenwirkungen verursachen. Wenn Sie bestimmte Arzneimittel zusammen mit SUSTIVA einnehmen, kann die Menge an SUSTIVA 600 mg in Ihrem Körper zu niedrig sein und es kann möglicherweise nicht dazu beitragen, Ihre HIV-Infektion zu kontrollieren. Das HIV-Virus in Ihrem Körper kann gegen SUSTIVA oder andere ähnliche HIV-Arzneimittel resistent werden.
Sie sollten SUSTIVA 600 mg nicht einnehmen, wenn Sie ATRIPLA einnehmen (Efavirenz, Emtricitabin, Tenofovirdisoproxilfumarat), es sei denn, Ihr Arzt hat es Ihnen gesagt.
Informieren Sie Ihren Arzt und Apotheker über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und Kräuterergänzungen. Einige Arzneimittel interagieren mit SUSTIVA.
Führen Sie eine Liste Ihrer Arzneimittel, um sie Ihrem Arzt und Apotheker zu zeigen.
Wie soll ich SUSTIVA einnehmen?
Wie und wann ist SUSTIVA einzunehmen?
Welche Nebenwirkungen kann SUSTIVA haben?
SUSTIVA 600 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Wenn Sie unter Schwindel, Konzentrationsschwierigkeiten oder Benommenheit leiden, fahren Sie kein Auto, bedienen Sie keine Maschinen und tun Sie nichts, bei dem Sie wachsam sein müssen.
Einige Symptome des Nervensystems (z. B. Verwirrtheit, verlangsamte Gedanken und körperliche Bewegungen sowie Wahnvorstellungen [falsche Überzeugungen] oder Halluzinationen [Sehen oder Hören von Dingen, die andere nicht sehen oder hören]) können Monate bis Jahre nach Beginn der SUSTIVA 600 mg-Therapie auftreten. Wenden Sie sich umgehend an Ihren Arzt, wenn eines dieser Symptome auftritt.
Informieren Sie sofort Ihren Arzt, wenn bei Ihnen eines der folgenden Symptome auftritt:
Zu den häufigsten Nebenwirkungen von SUSTIVA 600 mg gehören:
Bei einigen Patienten, die SUSTIVA einnahmen, traten erhöhte Blutfettwerte (Cholesterin und Triglyceride) auf. Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von SUSTIVA. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist SUSTIVA aufzubewahren?
Bewahren Sie SUSTIVA und alle Arzneimittel für Kinder unzugänglich auf.
Allgemeine Informationen zu SUSTIVA
Arzneimittel werden manchmal zu anderen als den in der Packungsbeilage aufgeführten Zwecken verschrieben. Verwenden Sie SUSTIVA 600 mg nicht für eine Erkrankung, für die es nicht verschrieben wurde. Geben Sie SUSTIVA 600 mg nicht an andere Personen, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Wenn Sie weitere Informationen wünschen, sprechen Sie mit Ihrem Arzt. Sie können Ihren Apotheker oder Arzt um Informationen über SUSTIVA bitten, die für Angehörige der Gesundheitsberufe bestimmt sind. Weitere Informationen erhalten Sie unter www.sustiva.com oder telefonisch unter 1-800-321-1335.
Welche Inhaltsstoffe enthält SUSTIVA?
Wirkstoff: Efavirenz
Inaktive Zutaten:
SUSTIVA 600 mg Kapseln: Lactosemonohydrat, Magnesiumstearat, Natriumlaurylsulfat und Natriumstärkeglykolat. Die Kapselhülle enthält Gelatine, Natriumlaurylsulfat, Titandioxid und/oder gelbes Eisenoxid. Die Kapselhülle kann auch Siliziumdioxid enthalten. Die Kapseln sind mit Tinte bedruckt, die Carmine 40 Blue, FD&C Blue No. 2 und Titandioxid enthält.
SUSTIVA-Tabletten: Croscarmellose-Natrium, Hydroxypropylcellulose, Lactosemonohydrat, Magnesiumstearat, mikrokristalline Cellulose und Natriumlaurylsulfat. Die Filmbeschichtung der Tablette enthält Opadry Yellow und Opadry Clear. Die Tabletten sind mit Carnaubawachs poliert und mit violetter Tinte, Opacode WB, bedruckt.
Diese Patienteninformation wurde von der US Food and Drug Administration genehmigt.
Gebrauchsanweisung
SUSTIVA® (sus-TEE-vah) (Efavirenz) Kapseln
Zubereitung einer Dosis von SUSTIVA 600 mg nach der Kapselstreumethode
Lesen Sie diese Gebrauchsanweisung, bevor Sie Ihre erste Dosis SUSTIVA gemischt mit Nahrung oder Säuglingsanfangsnahrung nach der Kapselstreumethode zubereiten, jedes Mal, wenn Sie eine Nachfüllung erhalten, und bei Bedarf. Möglicherweise gibt es neue Informationen. Diese Informationen ersetzen nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung. Wenden Sie sich an Ihren Arzt oder Apotheker, wenn Sie Fragen zum Mischen oder Verabreichen einer Dosis von SUSTIVA 200 mg nach der Kapselsprühmethode haben.
Wichtige Informationen:
Zubereitung einer Dosis von SUSTIVA 600 mg gemischt mit Nahrung nach der Kapselstreumethode.
Bevor Sie eine Dosis von SUSTIVA 200 mg gemischt mit Nahrung nach der Kapselstreumethode zubereiten, besorgen Sie die folgenden Vorräte:
Schritt 1. Wählen Sie eine saubere, ebene Arbeitsfläche. Legen Sie ein sauberes Papiertuch auf die Arbeitsfläche. Legen Sie dann die anderen Vorräte auf das Papiertuch.
Schritt 2. Waschen und trocknen Sie Ihre Hände gut.
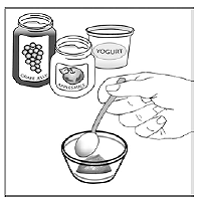
Schritt 3. Geben Sie 1 bis 2 Teelöffel weiche Lebensmittel wie Apfelmus, Traubengelee oder Joghurt in den kleinen Behälter (siehe Abbildung A ). Die Farbe und Dicke des Lebensmittels kann sich ändern, wenn es mit dem Arzneimittel gemischt wird.
Abbildung A
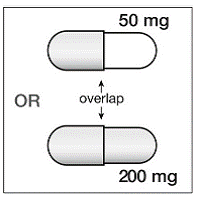
Schritt 4. Es gibt 2 Teile der SUSTIVA 200 mg Kapsel. Sehen Sie sich die SUSTIVA-Kapsel an, um zu sehen, welcher Teil der Kapsel den anderen Teil überlappt (siehe Abbildung B ).
Abbildung B
Schritt 5. Halten Sie die SUSTIVA-Kapsel seitlich (waagerecht) direkt über den Lebensmittelbehälter. Halten Sie jedes Ende der SUSTIVA-Kapsel zwischen Daumen und Zeigefinger (siehe Abbildung C ).
Abbildung C
Schritt 6. Verwenden Sie Ihren Daumen und Zeigefinger, um nahe dem Ende des überlappenden Teils der SUSTIVA 600-mg-Kapsel zusammenzudrücken (siehe Abbildung D ).
Abbildung D
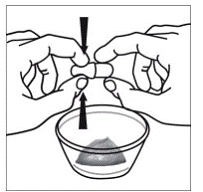
Drehen Sie dann beide Enden der SUSTIVA 600 mg-Kapsel vorsichtig in entgegengesetzte Richtungen, um sie zu öffnen (siehe Abbildung E ). Achten Sie darauf, den Kapselinhalt nicht zu verschütten oder in der Luft zu verteilen.
Abbildung E

Schritt 7. Streuen Sie den Inhalt der SUSTIVA-Kapsel auf das Futter (siehe Abbildung F ).
Abbildung F

Wenn die verschriebene Gesamtdosis mehr als 1 Kapsel beträgt, befolgen Sie die Schritte 4 bis 7 für jede Kapsel. Fügen Sie keine weiteren Lebensmittel hinzu.
Die Schritte 8 bis 11 sollten abgeschlossen sein innerhalb von 30 Minuten des Mischens der Medizin (siehe Abbildung G ).
Abbildung G

Schritt 8. Verwenden Sie den kleinen Löffel, um den Kapselinhalt und die Nahrung vorsichtig miteinander zu vermischen (siehe Abbildung H ). Streusel lösen sich nicht auf. Die Mischung sieht körnig aus, sollte aber nicht klumpig sein.
Abbildung H
Schritt 9. Verwenden Sie den kleinen Löffel, um die Mischung aus Nahrung und Kapselinhalt zu verabreichen oder einzunehmen. Stellen Sie sicher, dass die gesamte Mischung geschluckt wird.
Schritt 10. Geben Sie weitere 2 Teelöffel der Nahrung in den leeren Behälter und rühren Sie vorsichtig mit dem kleinen Löffel um, um sich mit dem eventuell noch im Behälter befindlichen Kapselinhalt zu vermischen.
Schritt 11. Verwenden Sie den kleinen Löffel, um die Mischung aus Nahrung und Kapselinhalt zu verabreichen oder einzunehmen. Stellen Sie sicher, dass die gesamte Mischung geschluckt wird.
Schritt 12. Behälter und Löffel waschen. Werfen Sie das Papiertuch weg und reinigen Sie die Arbeitsfläche. Wasche deine Hände.
Zubereitung einer Dosis von SUSTIVA 600 mg gemischt mit Säuglingsanfangsnahrung nach der Kapselstreumethode
Um sicherzustellen, dass Ihr Baby das gesamte Arzneimittel erhält, geben Sie Ihrem Baby den Inhalt der SUSTIVA-Kapsel nicht in einer Flasche.
Bevor Sie eine Dosis SUSTIVA gemischt mit Säuglingsanfangsnahrung nach der Kapselstreumethode zubereiten, besorgen Sie die folgenden Vorräte:
Abbildung I
Schritt 1. Bereiten Sie die Säuglingsnahrung gemäß den Anweisungen auf der Packung der Säuglingsnahrung zu. Sie werden etwa 1 Unze der Formel verwenden, um das Arzneimittel zu verabreichen. Restliche Formel sollte dem Kind 2 Stunden lang nicht gegeben werden.
Schritt 2. Wählen Sie eine saubere, ebene Arbeitsfläche. Legen Sie ein sauberes Papiertuch auf die Arbeitsfläche. Legen Sie die Materialien, die Sie benötigen, auf das Papiertuch.
Schritt 3. Waschen und trocknen Sie Ihre Hände gut.
Schritt 4. Gießen Sie 2 Teelöffel zimmerwarme Säuglingsnahrung in den Behälter (siehe Abbildung J ).
Abbildung J
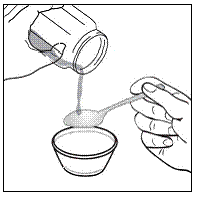
Schritt 5. Die SUSTIVA-Kapsel besteht aus 2 Teilen. Sehen Sie sich die SUSTIVA-Kapsel an, um zu sehen, welcher Teil der Kapsel den anderen Teil überlappt (siehe Abbildung K ).
Abbildung K
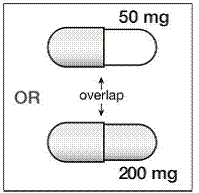
Schritt 6. Halten Sie die SUSTIVA-Kapsel seitlich (waagerecht) direkt über das Behältnis mit der Säuglingsanfangsnahrung. Halten Sie jedes Ende der SUSTIVA 600 mg-Kapsel zwischen Daumen und Zeigefinger (siehe Abbildung L ).
Abbildung L
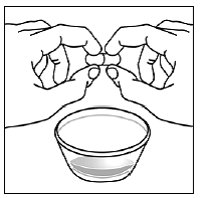
Schritt 7. Drücken Sie mit Daumen und Zeigefinger nahe dem Ende des überlappenden Teils der SUSTIVA-Kapsel zusammen (siehe Abbildung M ).
Abbildung M
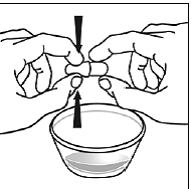
Drehen Sie dann beide Enden der SUSTIVA-Kapsel vorsichtig in entgegengesetzte Richtungen, um sie zu öffnen (siehe Abbildung N ). Achten Sie darauf, den Kapselinhalt nicht zu verschütten oder in der Luft zu verteilen.
Abbildung N
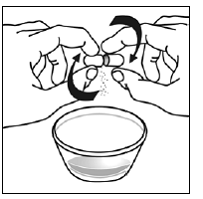
Schritt 8. Streuen Sie den Inhalt der SUSTIVA 600 mg Kapsel auf die Säuglingsnahrung (siehe Abbildung O ).
Abbildung O
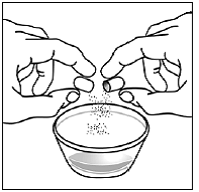
Wenn die verschriebene Gesamtdosis mehr als 1 Kapsel beträgt, befolgen Sie die Schritte 5 bis 8 für jede Kapsel. Fügen Sie keine weitere Säuglingsnahrung hinzu.
Die Schritte 9 bis 12 sollten abgeschlossen sein innerhalb von 30 Minuten des Mischens der Medizin (siehe Abbildung P ).
Abbildung P

Schritt 9. Halten Sie den Behälter mit einer Hand. Verwenden Sie mit der anderen Hand den Teelöffel, um den Kapselinhalt und die Säuglingsnahrung vorsichtig zu vermischen (siehe Abbildung Q ). Streusel lösen sich nicht auf. Die Mischung sieht körnig aus, sollte aber nicht klumpig sein.
Abbildung Q
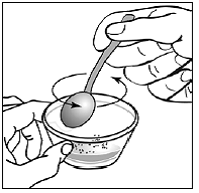
Schritt 10. So ziehen Sie die gesamte Mischung in die Applikationsspritze für Zubereitungen zum Einnehmen auf:
Abbildung R
Abbildung S
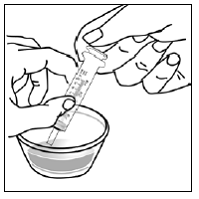
Abbildung T
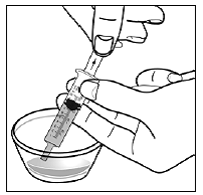
Schritt 11. Setzen Sie die Spitze der Spritze in den Mund Ihres Babys entlang der inneren Wange (siehe Abbildung u ). Drücken Sie langsam auf den Kolben, um Ihrem Baby die gesamte Mischung zu geben.
Abbildung u
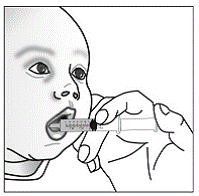
Schritt 12. Um sicherzustellen, dass Ihr Baby das gesamte Arzneimittel erhält:
Abbildung V
Schritt 13. Entfernen Sie den Kolben von der Applikationsspritze für Zubereitungen zum Einnehmen. Waschen Sie das Behältnis, den Teelöffel und die orale Dosierspritze. Lassen Sie den Kolben und den Spritzenzylinder trocknen, bevor Sie sie wieder zusammensetzen.
Schritt 14. Werfen Sie das Papiertuch weg und reinigen Sie die Arbeitsfläche. Wasche deine Hände.
Wie soll ich SUSTIVA 600 mg Kapseln aufbewahren?
Bewahren Sie SUSTIVA 600 mg Kapseln und alle Arzneimittel außerhalb der Reichweite von Kindern auf.
Diese Gebrauchsanweisung wurde von der US Food and Drug Administration genehmigt.