Arimidex 1mg Anastrozole Verwendung, Nebenwirkungen, Stärke und Dosierung. Preis in Online-Apotheke. Generika medikamente rezeptfrei.
Was ist Arimidex und wie wird es angewendet?
Arimidex 1 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Brustkrebs bei postmenopausalen Frauen. Arimidex kann allein oder mit anderen Medikamenten verwendet werden.
Arimidex gehört zu einer Klasse von Medikamenten namens Antineoplastika, Aromatasehemmer.
Es ist nicht bekannt, ob Arimidex bei Kindern sicher und wirksam ist.
Welche Nebenwirkungen kann Arimidex haben?
Arimidex kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Arimidex gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Arimidex. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
ARIMIDEX (Anastrozol)-Tabletten zur oralen Verabreichung enthalten 1 mg Anastrozol, einen nichtsteroidalen Aromatasehemmer. Es wird chemisch als 1,3-Benzendiacetonitril, a, a, a', a'-tetramethyl-5-(1H-1,2,4-triazol-1-ylmethyl) beschrieben. Seine Summenformel ist C17H19N5 und seine Strukturformel ist:
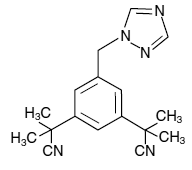
Anastrozol ist ein cremefarbenes Pulver mit einem Molekulargewicht von 293,4. Anastrozol hat eine mäßige Wasserlöslichkeit (0,5 mg/ml bei 25 °C); die Löslichkeit ist im physiologischen Bereich pH-unabhängig. Anastrozol ist in Methanol, Aceton, Ethanol und Tetrahydrofuran frei löslich und in Acetonitril sehr gut löslich.
Jede Tablette enthält als inaktive Bestandteile: Lactose, Magnesiumstearat, Hydroxypropylmethylcellulose, Polyethylenglykol, Povidon, Natriumstärkeglykolat und Titandioxid.
INDIKATIONEN
Adjuvante Behandlung
ARIMIDEX ist indiziert zur adjuvanten Behandlung von postmenopausalen Frauen mit hormonrezeptorpositivem Brustkrebs im Frühstadium.
First-Line-Behandlung
ARIMIDEX 1 mg ist angezeigt für die Erstlinienbehandlung von postmenopausalen Frauen mit hormonrezeptorpositivem oder hormonrezeptorunbekanntem lokal fortgeschrittenem oder metastasiertem Brustkrebs.
Second-Line-Behandlung
ARIMIDEX 1 mg ist indiziert zur Behandlung von fortgeschrittenem Brustkrebs bei postmenopausalen Frauen mit Krankheitsprogression nach einer Tamoxifen-Therapie. Patienten mit ER-negativer Erkrankung und Patienten, die auf eine vorangegangene Tamoxifen-Therapie nicht ansprachen, sprachen selten auf ARIMIDEX an.
DOSIERUNG UND ANWENDUNG
Empfohlene Dosis
Die Dosis von ARIMIDEX beträgt eine 1-mg-Tablette, die einmal täglich eingenommen wird. Bei Patientinnen mit fortgeschrittenem Brustkrebs sollte ARIMIDEX 1 mg bis zur Tumorprogression fortgesetzt werden. ARIMIDEX kann mit oder ohne Nahrung eingenommen werden.
Für die adjuvante Behandlung von Brustkrebs im Frühstadium bei postmenopausalen Frauen ist die optimale Therapiedauer nicht bekannt. In der ATAC-Studie wurde ARIMIDEX 1 mg fünf Jahre lang verabreicht [siehe Klinische Studien ].
Bei Patienten mit eingeschränkter Nierenfunktion oder bei älteren Patienten ist keine Dosisanpassung erforderlich [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Patienten mit eingeschränkter Leberfunktion
Für Patienten mit leichter bis mittelschwerer Leberfunktionsstörung werden keine Dosisänderungen empfohlen. ARIMIDEX wurde bei Patienten mit schwerer Leberfunktionsstörung nicht untersucht [siehe Verwendung in bestimmten Populationen ].
WIE GELIEFERT
Darreichungsformen und Stärken
Die Tabletten sind weiß, bikonvex, filmbeschichtet und enthalten 1 mg Anastrozol. Die Tabletten sind auf einer Seite mit einem Logo bedruckt, das aus einem Buchstaben „A“ (Großbuchstabe) mit einer Pfeilspitze besteht, die am Fuß des verlängerten rechten Beins des „A“ befestigt ist, und auf der Rückseite mit der Tablettenstärkemarkierung „Adx 1 “.
Lagerung und Handhabung
Diese Tabletten sind in Flaschen mit 30 Tabletten ( NDC 0310-0201-30).
Lagerung
Bei kontrollierter Raumtemperatur lagern, 20-25°C (68-77°F) [siehe USP ].
Vertrieb durch: AstraZeneca Pharmaceuticals LP Wilmington, DE 19850. Überarbeitet: Mai 2014
NEBENWIRKUNGEN
Schwerwiegende Nebenwirkungen von ARIMIDEX, die bei weniger als 1 von 10.000 Patienten auftreten, sind: 1) Hautreaktionen wie Läsionen, Geschwüre oder Blasen; 2) allergische Reaktionen mit Schwellung von Gesicht, Lippen, Zunge und/oder Rachen. Dies kann zu Schluck- und/oder Atembeschwerden führen; und 3) Veränderungen bei Bluttests der Leberfunktion, einschließlich Leberentzündung mit Symptomen, die ein allgemeines Unwohlsein umfassen können, mit oder ohne Gelbsucht, Leberschmerzen oder Leberschwellung.
Häufige Nebenwirkungen (mit einer Häufigkeit von ≥ 10 %) bei Frauen, die ARIMIDEX 1 mg einnahmen, waren: Hitzewallungen, Asthenie, Arthritis, Schmerzen, Arthralgie, Bluthochdruck, Depression, Übelkeit und Erbrechen, Hautausschlag, Osteoporose, Knochenbrüche, Rückenschmerzen, Schlaflosigkeit, Kopfschmerzen, Knochenschmerzen, periphere Ödeme, verstärkter Husten, Dyspnoe, Pharyngitis und Lymphödem.
In der ATAC-Studie waren Hitzewallungen die am häufigsten berichtete Nebenwirkung (> 0,1 %), die zu einem Therapieabbruch führte, obwohl es in der ARIMIDEX-1-mg-Gruppe weniger Patienten gab, die die Therapie aufgrund von Hitzewallungen abbrachen.
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Erfahrung mit klinischen Studien
Adjuvante Therapie
Die Daten zu Nebenwirkungen für die adjuvante Therapie basieren auf der ATAC-Studie [siehe Klinische Studien ]. Die mediane Dauer der adjuvanten Behandlung zur Sicherheitsbewertung betrug 59,8 Monate bzw. 59,6 Monate bei Patienten, die ARIMIDEX 1 mg bzw. Tamoxifen 20 mg erhielten.
Nebenwirkungen, die in jeder Behandlungsgruppe während der Behandlung oder innerhalb von 14 Tagen nach Behandlungsende mit einer Inzidenz von mindestens 5 % auftraten, sind in Tabelle 1 aufgeführt.
Bestimmte Nebenwirkungen und Kombinationen von Nebenwirkungen wurden prospektiv zur Analyse spezifiziert, basierend auf den bekannten pharmakologischen Eigenschaften und Nebenwirkungsprofilen der beiden Arzneimittel (siehe Tabelle 2).
Ischämische kardiovaskuläre Ereignisse
Zwischen den Behandlungsarmen in der Gesamtpopulation von 6186 Patienten gab es keinen statistischen Unterschied bei ischämischen kardiovaskulären Ereignissen (4 % ARIMIDEX vs. 3 % Tamoxifen).
In der Gesamtpopulation wurde Angina pectoris bei 71/3092 (2,3 %) Patienten im Arm mit ARIMIDEX 1 mg und bei 51/3094 (1,6 %) Patienten im Arm mit Tamoxifen berichtet; Myokardinfarkt wurde bei 37/3092 (1,2 %) Patienten im Arm mit ARIMIDEX 1 mg und bei 34/3094 (1,1 %) Patienten im Arm mit Tamoxifen berichtet.
Bei Frauen mit vorbestehender ischämischer Herzkrankheit 465/6186 (7,5 %) betrug die Inzidenz ischämischer kardiovaskulärer Ereignisse 17 % bei Patientinnen unter ARIMIDEX und 10 % bei Patientinnen unter Tamoxifen. In dieser Patientenpopulation wurde Angina pectoris bei 25/216 (11,6 %) Patienten, die ARIMIDEX erhielten, und bei 13/249 (5,2 %) Patienten, die Tamoxifen erhielten, berichtet; Myokardinfarkt wurde bei 2/216 (0,9 %) Patienten, die ARIMIDEX erhielten, und bei 8/249 (3,2 %) Patienten, die Tamoxifen erhielten, berichtet.
Ergebnisse der Knochenmineraldichte
Die Ergebnisse der Knochensubstudie der ATAC-Studie nach 12 und 24 Monaten zeigten, dass Patienten, die ARIMIDEX 1 mg erhielten, im Vergleich zum Ausgangswert eine mittlere Abnahme sowohl der Lendenwirbelsäule als auch der Knochenmineraldichte (BMD) der gesamten Hüfte aufwiesen. Patienten, die Tamoxifen erhielten, hatten im Vergleich zum Ausgangswert einen mittleren Anstieg sowohl der Lendenwirbelsäule als auch der gesamten Hüft-BMD.
Da ARIMIDEX den zirkulierenden Östrogenspiegel senkt, kann es zu einer Verringerung der Knochenmineraldichte führen.
In einer Post-Marketing-Studie wurden die kombinierten Wirkungen von ARIMIDEX 1 mg und dem Bisphosphonat-Risedronat auf Veränderungen der BMD gegenüber dem Ausgangswert und Marker für Knochenresorption und -bildung bei postmenopausalen Frauen mit Hormonrezeptor-positivem Brustkrebs im Frühstadium untersucht. Alle Patienten erhielten eine Kalzium- und Vitamin-D-Ergänzung. Nach 12 Monaten wurde bei Patienten, die keine Bisphosphonate erhielten, eine geringe Verringerung der Knochenmineraldichte der Lendenwirbelsäule festgestellt. Die Behandlung mit Bisphosphonaten bewahrte die Knochendichte bei den meisten Patienten mit Frakturrisiko.
Postmenopausale Frauen mit Brustkrebs im Frühstadium, die mit ARIMIDEX 1 mg behandelt werden sollen, sollten ihren Knochenstatus gemäß den Behandlungsrichtlinien kontrollieren lassen, die bereits für postmenopausale Frauen mit ähnlichem Risiko für Fragilitätsfrakturen verfügbar sind.
Cholesterin
Während der ATAC-Studie wurde von mehr Patienten, die ARIMIDEX 1 mg erhielten, im Vergleich zu Patienten, die Tamoxifen erhielten, ein erhöhtes Serumcholesterin berichtet (9 % gegenüber 3,5 %).
In einer Post-Marketing-Studie wurden auch mögliche Auswirkungen von ARIMIDEX auf das Lipidprofil untersucht. In der primären Analysepopulation für Lipide (ARIMIDEX 1 mg allein) gab es keine klinisch signifikante Veränderung des LDL-C vom Ausgangswert bis zum 12. Monat und des HDL-C vom Ausgangswert bis zum 12. Monat.
In der Sekundärpopulation für Lipide (ARIMIDEX + Risedronat) gab es ebenfalls keine klinisch signifikante Veränderung von LDL-C und HDL-C vom Ausgangswert bis zu 12 Monaten.
In beiden Lipidpopulationen gab es nach 12 Monaten im Vergleich zum Ausgangswert keinen klinisch signifikanten Unterschied beim Gesamtcholesterin (TC) oder den Serumtriglyceriden (TG).
In dieser Studie hatte eine 12-monatige Behandlung mit ARIMIDEX 1 mg allein eine neutrale Wirkung auf das Lipidprofil. Die Kombinationsbehandlung mit ARIMIDEX und Risedronat hatte ebenfalls eine neutrale Wirkung auf das Lipidprofil.
Die Studie liefert Beweise dafür, dass postmenopausale Frauen mit Brustkrebs im Frühstadium, die mit ARIMIDEX 1 mg behandelt werden sollen, gemäß den aktuellen Richtlinien des National Cholesterol Education Program für das kardiovaskuläre risikobasierte Management einzelner Patienten mit LDL-Erhöhungen behandelt werden sollten.
Andere Nebenwirkungen
Patienten, die ARIMIDEX erhielten, hatten im Vergleich zu Patienten, die Tamoxifen erhielten, eine Zunahme von Gelenkerkrankungen (einschließlich Arthritis, Arthrose und Arthralgie). Patienten, die ARIMIDEX erhielten, hatten eine erhöhte Inzidenz aller Frakturen (insbesondere Frakturen der Wirbelsäule, der Hüfte und des Handgelenks) [315 (10 %)] im Vergleich zu Patienten, die Tamoxifen erhielten [209 (7 %)].
Patienten, die ARIMIDEX 1 mg erhielten, hatten eine höhere Inzidenz des Karpaltunnelsyndroms [78 (2,5 %)] im Vergleich zu Patienten, die Tamoxifen erhielten [22 (0,7 %)].
Vaginale Blutungen traten bei den mit Tamoxifen behandelten Patientinnen häufiger auf als bei den mit ARIMIDEX behandelten Patientinnen, 317 (10 %) gegenüber 167 (5 %).
Patienten, die ARIMIDEX erhielten, hatten im Vergleich zu Patienten, die Tamoxifen erhielten, eine geringere Inzidenz von Hitzewallungen, Vaginalblutungen, Vaginalausfluss, Endometriumkarzinom, venösen thromboembolischen Ereignissen und ischämischen zerebrovaskulären Ereignissen.
10-Jahres-Median-Follow-up-Sicherheitsergebnisse aus der ATAC-Studie
Die Ergebnisse stimmen mit den vorherigen Analysen überein.
Schwerwiegende Nebenwirkungen waren bei ARIMIDEX (50 %) und Tamoxifen (51 %) ähnlich.
First-Line-Therapie
Nebenwirkungen, die mit einer Inzidenz von mindestens 5 % in beiden Behandlungsgruppen der Studien 0030 und 0027 während oder innerhalb von 2 Wochen nach Behandlungsende auftraten, sind in Tabelle 3 aufgeführt.
Weniger häufig berichtete Nebenwirkungen bei Patienten, die entweder in Studie 0030 oder in Studie 0027 ARIMIDEX 1 mg l mg erhielten, waren ähnlich denen, die für die Zweitlinientherapie berichtet wurden.
Basierend auf den Ergebnissen der Zweitlinientherapie und dem etablierten Sicherheitsprofil von Tamoxifen wurden die Inzidenzen von 9 vordefinierten Kategorien unerwünschter Ereignisse, die aufgrund ihrer Pharmakologie möglicherweise in ursächlichem Zusammenhang mit einer oder beiden Therapien stehen, statistisch analysiert. Zwischen den Behandlungsgruppen wurden keine signifikanten Unterschiede festgestellt.
Second-Line-Therapie
ARIMIDEX 1 mg wurde in zwei kontrollierten klinischen Studien (dh Studien 0004 und 0005) vertragen, wobei weniger als 3,3 % der mit ARIMIDEX behandelten Patienten und 4,0 % der mit Megestrolacetat behandelten Patienten die Studie aufgrund einer Nebenwirkung abbrachen.
Die Hauptnebenwirkung, die bei ARIMIDEX häufiger auftrat als bei Megestrolacetat, war Durchfall. Nebenwirkungen, die unabhängig von der Kausalität bei mehr als 5 % der Patienten in einer der Behandlungsgruppen in diesen beiden kontrollierten klinischen Studien berichtet wurden, sind nachstehend aufgeführt:
Körper als Ganzes: Grippesyndrom; Fieber; Nackenschmerzen; Unwohlsein; Unfallverletzung; Infektion
Herz-Kreislauf: Hypertonie; Thrombophlebitis
Leber: Gamma-GT erhöht; SGOT erhöht; SGPT erhöht Hämatologisch: Anämie; Leukopenie
Stoffwechsel und Ernährung: Alkalische Phosphatase erhöht; Gewichtsverlust
Die mittleren Gesamtcholesterinspiegel im Serum stiegen bei Patienten, die ARIMIDEX erhielten, um 0,5 mmol/l. Es wurde gezeigt, dass ein Anstieg des LDL-Cholesterins zu diesen Veränderungen beiträgt.
Bewegungsapparat: Myalgie; Arthralgie; pathologische Fraktur
Nervös: Schläfrigkeit; Verwirrtheit; Schlaflosigkeit; Angst; Nervosität
Atmung: Nebenhöhlenentzündung; Bronchitis; Schnupfen
Haut und Anhängsel: Haarausfall (Alopezie); Juckreiz
Urogenital: Infektion der Harnwege; Brustschmerzen
Die Inzidenzen der folgenden Nebenwirkungsgruppen, die aufgrund ihrer Pharmakologie möglicherweise in ursächlichem Zusammenhang mit einer oder beiden Therapien stehen, wurden statistisch analysiert: Gewichtszunahme, Ödeme, thromboembolische Erkrankungen, gastrointestinale Störungen, Hitzewallungen und vaginale Trockenheit. Diese sechs Gruppen und die in den Gruppen erfassten Nebenwirkungen wurden prospektiv definiert. Die Ergebnisse sind in der nachstehenden Tabelle gezeigt.
Post-Marketing-Erfahrung
Diese Nebenwirkungen werden freiwillig von einer Population ungewisser Größe gemeldet. Daher ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen. Folgendes wurde bei der Anwendung von Arimidex nach der Zulassung berichtet:
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Tamoxifen
Die gleichzeitige Anwendung von Anastrozol und Tamoxifen bei Brustkrebspatientinnen verringerte die Plasmakonzentration von Anastrozol um 27 %. Die gleichzeitige Verabreichung von Anastrozol und Tamoxifen hatte jedoch keinen Einfluss auf die Pharmakokinetik von Tamoxifen oder N-Desmethyltamoxifen. Bei einer medianen Nachbeobachtungszeit von 33 Monaten zeigte die Kombination von ARIMIDEX und Tamoxifen keinen Wirksamkeitsvorteil im Vergleich zu Tamoxifen bei allen Patienten sowie in der Hormonrezeptor-positiven Subpopulation. Dieser Behandlungsarm wurde aus der Studie ausgeschlossen [siehe Klinische Studien ]. Basierend auf den klinischen und pharmakokinetischen Ergebnissen der ATAC-Studie sollte Tamoxifen nicht zusammen mit Anastrozol verabreicht werden.
Östrogen
Östrogenhaltige Therapien sollten nicht zusammen mit ARIMIDEX 1 mg angewendet werden, da sie die pharmakologische Wirkung verringern können.
Warfarin
In einer Studie mit 16 männlichen Probanden veränderte Anastrozol weder die Exposition (gemessen anhand von Cmax und AUC) noch die antikoagulatorische Aktivität (gemessen anhand der Prothrombinzeit, der aktivierten partiellen Thromboplastinzeit und der Thrombinzeit) von R- und S- Warfarin.
Cytochrom P450 Basierend auf In-vitro- und In-vivo-Ergebnissen ist es unwahrscheinlich, dass die gleichzeitige Verabreichung von ARIMIDEX 1 mg andere Arzneimittel aufgrund der Hemmung von Cytochrom P450 beeinflusst [siehe KLINISCHE PHARMAKOLOGIE ].
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Ischämische kardiovaskuläre Ereignisse
Bei Frauen mit vorbestehender ischämischer Herzkrankheit wurde in der ATAC-Studie unter ARIMIDEX eine erhöhte Inzidenz ischämischer kardiovaskulärer Ereignisse beobachtet (17 % der Patientinnen unter ARIMIDEX 1 mg und 10 % der Patientinnen unter Tamoxifen). Risiko und Nutzen einer ARIMIDEX-Therapie bei Patienten mit vorbestehender ischämischer Herzkrankheit abwägen [siehe NEBENWIRKUNGEN ]
Knocheneffekte
Die Ergebnisse der Knochensubstudie der ATAC-Studie nach 12 und 24 Monaten zeigten, dass Patienten, die ARIMIDEX erhielten, im Vergleich zum Ausgangswert eine mittlere Abnahme sowohl der Lendenwirbelsäulen- als auch der gesamten Hüftknochenmineraldichte (BMD) aufwiesen. Patienten, die Tamoxifen erhielten, hatten im Vergleich zum Ausgangswert einen mittleren Anstieg sowohl der Lendenwirbelsäule als auch der gesamten Hüft-BMD. Erwägen Sie eine Überwachung der Knochenmineraldichte bei Patienten, die mit ARIMIDEX behandelt werden [siehe NEBENWIRKUNGEN ].
Cholesterin
Während der ATAC-Studie wurde von mehr Patienten, die ARIMIDEX 1 mg erhielten, im Vergleich zu Patienten, die Tamoxifen erhielten, ein erhöhter Serumcholesterinspiegel berichtet (9 % gegenüber 3,5 %) [siehe NEBENWIRKUNGEN ].
Informationen zur Patientenberatung
Sehen Von der FDA zugelassene Patientenkennzeichnung (PATIENT INFORMATION).
Schwangerschaft
Die Patienten sollten darauf hingewiesen werden, dass ARIMIDEX 1 mg den Fötus schädigen kann. Sie sollten auch darauf hingewiesen werden, dass ARIMIDEX 1 mg nicht zur Anwendung bei prämenopausalen Frauen bestimmt ist; Daher sollten sie, wenn sie schwanger werden, die Einnahme von ARIMIDEX abbrechen und unverzüglich ihren Arzt kontaktieren.
Allergische (Überempfindlichkeits-) Reaktionen
Die Patienten sollten über die Möglichkeit schwerwiegender allergischer Reaktionen mit Schwellungen von Gesicht, Lippen, Zunge und/oder Rachen (Angioödem), die Schluck- und/oder Atembeschwerden verursachen können, informiert werden und unverzüglich einen Arzt aufsuchen.
Ischämische kardiovaskuläre Ereignisse
Patienten mit vorbestehender ischämischer Herzkrankheit sollten darüber informiert werden, dass bei der Anwendung von ARIMIDEX 1 mg im Vergleich zur Anwendung von Tamoxifen eine erhöhte Inzidenz kardiovaskulärer Ereignisse beobachtet wurde. Wenn Patienten neue oder sich verschlechternde Brustschmerzen oder Kurzatmigkeit haben, sollten sie sofort einen Arzt aufsuchen.
Knocheneffekte
Die Patienten sollten darüber informiert werden, dass ARIMIDEX den Östrogenspiegel senkt. Dies kann zu einem Verlust des Mineralgehalts der Knochen führen, was die Knochenfestigkeit verringern kann. Eine mögliche Folge eines verminderten Mineralgehalts der Knochen ist ein erhöhtes Frakturrisiko.
Cholesterin
Die Patienten sollten darüber informiert werden, dass während der Behandlung mit ARIMIDEX ein erhöhter Cholesterinspiegel beobachtet werden kann.
Kitzeln, Kribbeln oder Taubheit
Die Patienten sollten darüber informiert werden, dass sie ihren Arzt benachrichtigen sollten, wenn sie ein Kitzeln, Kribbeln oder Taubheitsgefühl verspüren.
Tamoxifen
Die Patienten sollten darauf hingewiesen werden, ARIMIDEX nicht zusammen mit Tamoxifen einzunehmen.
Verpasste Dosen
Informieren Sie die Patienten, dass sie eine vergessene Dosis nachholen, sobald sie sich daran erinnern. Wenn es fast Zeit für die nächste Dosis ist, überspringen Sie die vergessene Dosis und nehmen Sie die nächste reguläre Dosis ein. Die Patienten sollten nicht zwei Dosen gleichzeitig einnehmen.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Eine konventionelle Karzinogenese-Studie an Ratten mit Dosen von 1,0 bis 25 mg/kg/Tag (etwa das 10- bis 243-fache der empfohlenen Tageshöchstdosis beim Menschen auf mg/m²-Basis), die bis zu 2 Jahre lang per Schlundsonde verabreicht wurden, zeigte einen Anstieg der Inzidenz von hepatozellulären Adenomen und Karzinomen und uterinen Stromapolypen bei Frauen und Schilddrüsenadenomen bei Männern bei hoher Dosis. Bei Frauen wurde ein dosisabhängiger Anstieg der Ovarial- und Uterushyperplasie beobachtet. Bei 25 mg/kg/Tag waren die Plasma-AUC0-24-Stunden-Werte bei Ratten 110- bis 125-mal höher als die Werte, die bei postmenopausalen Probanden bei der empfohlenen Dosis auftraten. Eine separate Kanzerogenitätsstudie an Mäusen mit oralen Dosen von 5 bis 50 mg/kg/Tag (etwa das 24- bis 243-fache der empfohlenen Tageshöchstdosis beim Menschen auf mg/m²-Basis) über einen Zeitraum von bis zu 2 Jahren ergab eine Zunahme der Inzidenz gutartiger Nebenwirkungen Stroma-, Epithel- und Granulosazelltumoren der Eierstöcke bei allen Dosierungen. Auch bei weiblichen Mäusen wurde eine dosisabhängige Zunahme der Ovarialhyperplasie beobachtet. Diese ovariellen Veränderungen gelten als nagetierspezifische Effekte der Aromatasehemmung und sind für den Menschen von fraglicher Bedeutung. Die Inzidenz von Lymphosarkomen war bei Männern und Frauen bei der hohen Dosis erhöht. Bei 50 mg/kg/Tag waren die Plasma-AUC-Spiegel bei Mäusen 35- bis 40-mal höher als bei postmenopausalen Probandinnen bei der empfohlenen Dosis.
ARIMIDEX 1 mg hat sich in In-vitro-Tests (Ames- und E. coli-Bakterientests, CHO-K1-Genmutationstest) oder in vitro (Chromosomenaberrationen in menschlichen Lymphozyten) oder in vivo (Mikronukleustest in Ratten) nicht als mutagen oder klastogen erwiesen. .
Die orale Verabreichung von Anastrozol an weibliche Ratten (von 2 Wochen vor der Paarung bis zum 7. Trächtigkeitstag) führte bei 1 mg/kg/Tag (etwa dem 10-fachen der beim Menschen empfohlenen Dosis auf mg/m²-Basis) zu einer signifikanten Inzidenz von Unfruchtbarkeit und einer verringerten Anzahl lebensfähiger Trächtigkeiten und 9-mal höher als die AUC0-24 h, die bei postmenopausalen Freiwilligen bei der empfohlenen Dosis gefunden wurde). Bei Dosen von mindestens 0,02 mg/kg/Tag (etwa ein Fünftel der empfohlenen Humandosis auf mg/m²-Basis) war der Präimplantationsverlust an Eizellen oder Fötus erhöht. Eine Wiederherstellung der Fertilität wurde nach einer 5-wöchigen Periode ohne Einnahme beobachtet, die auf eine 3-wöchige Einnahme folgte. Es ist nicht bekannt, ob diese bei weiblichen Ratten beobachteten Wirkungen auf eine beeinträchtigte Fertilität beim Menschen hindeuten.
Mehrfachdosisstudien an Ratten, denen Anastrozol über 6 Monate in Dosen von mindestens 1 mg/kg/Tag verabreicht wurde (was zu Cssmax und AUC 0-24 h von Anastrozol im Plasma führte, die 19- und 9-mal höher waren als die entsprechenden Werte, die in der Postmenopause gefunden wurden Probanden in der empfohlenen Dosis) führte zu einer Hypertrophie der Eierstöcke und dem Vorhandensein von follikulären Zysten. Darüber hinaus wurden in 6-monatigen Studien an Hündinnen, denen Dosen von mindestens 1 mg/kg/Tag verabreicht wurden, hyperplastische Uteri beobachtet (was zu Plasma-Anastrozol-Cssmax und -AUC0-24-Stunden führte, die 22-mal bzw. 16-mal höher waren als die entsprechenden Werte, die bei postmenopausalen Frauen bei der empfohlenen Dosis gefunden wurden). Es ist nicht bekannt, ob diese Wirkungen auf die Fortpflanzungsorgane von Tieren mit einer Beeinträchtigung der Fertilität bei Frauen vor der Menopause zusammenhängen.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschaftskategorie X [sehen KONTRAINDIKATIONEN ]
ARIMIDEX kann den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird, und bietet prämenopausalen Frauen mit Brustkrebs keinen klinischen Nutzen. ARIMIDEX ist kontraindiziert bei Frauen, die schwanger sind oder schwanger werden könnten. In Tierversuchen verursachte Anastrozol Schwangerschaftsversagen, erhöhten Schwangerschaftsverlust und Anzeichen einer verzögerten fötalen Entwicklung. Es liegen keine Studien zur Anwendung von ARIMIDEX 1 mg bei Schwangeren vor. Wenn ARIMIDEX 1 mg während der Schwangerschaft angewendet wird oder wenn die Patientin während der Einnahme dieses Arzneimittels schwanger wird, sollte die Patientin über die potenzielle Gefahr für den Fötus und das potenzielle Risiko eines Schwangerschaftsverlusts informiert werden.
In Reproduktionsstudien an Tieren erhielten trächtige Ratten und Kaninchen Anastrozol während der Organogenese in Dosen von mindestens 1 (Ratten) und 1/3 (Kaninchen) der empfohlenen Humandosis auf mg/m²-Basis. Bei beiden Spezies passierte Anastrozol die Plazentaschranke, und es kam zu einem erhöhten Schwangerschaftsverlust (erhöhter Prä- und/oder Postimplantationsverlust, erhöhte Resorption und verringerte Anzahl lebender Föten). Bei Ratten waren diese Wirkungen dosisabhängig und das Plazentagewicht war signifikant erhöht. Fetotoxizität, einschließlich verzögerter fötaler Entwicklung (d. h. unvollständige Ossifikation und vermindertes fetales Körpergewicht), trat bei Ratten bei Anastrozol-Dosen auf, die 19-mal höhere Plasmaspiegel als Serumspiegel beim Menschen bei der therapeutischen Dosis erzeugten (AUC 0-24 Std. 9-mal höher). . Bei Kaninchen verursachte Anastrozol bei Dosierungen, die mindestens dem 16-fachen der empfohlenen Humandosis auf mg/m²-Basis entsprachen, einen Schwangerschaftsausfall [siehe Tiertoxikologie und/oder Pharmakologie ].
Stillende Mutter
Es ist nicht bekannt, ob Anastrozol in die Muttermilch übergeht. Da viele Arzneimittel in die Muttermilch ausgeschieden werden und aufgrund der für Anastrozol in Tierstudien nachgewiesenen tumorigenen Wirkung oder des Potenzials schwerwiegender Nebenwirkungen bei gestillten Säuglingen sollte eine Entscheidung getroffen werden, ob das Stillen beendet oder das Arzneimittel abgesetzt werden soll Bedeutung des Medikaments für die Mutter.
Pädiatrische Verwendung
Klinische Studien an pädiatrischen Patienten umfassten eine placebokontrollierte Studie an pubertierenden Jungen im heranwachsenden Alter mit Gynäkomastie und eine einarmige Studie an Mädchen mit McCune-Albright-Syndrom und fortschreitender vorzeitiger Pubertät. Die Wirksamkeit von ARIMIDEX 1 mg bei der Behandlung der pubertären Gynäkomastie bei heranwachsenden Jungen und bei der Behandlung der vorzeitigen Pubertät bei Mädchen mit McCune-Albright-Syndrom wurde nicht nachgewiesen.
Gynäkomastie-Studie
In eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Studie wurden 80 Jungen mit pubertärer Gynäkomastie im Alter von 11 bis 18 Jahren aufgenommen. Die Patienten wurden randomisiert einer täglichen Behandlung mit entweder ARIMIDEX 1 mg oder Placebo zugeteilt. Nach 6-monatiger Behandlung gab es keinen statistisch signifikanten Unterschied im Prozentsatz der Patientinnen, bei denen eine Verringerung der Gynäkomastie um ≥ 50 % auftrat (primäre Wirksamkeitsanalyse). Sekundäre Wirksamkeitsanalysen (absolute Veränderung des Brustvolumens, Prozentsatz der Patientinnen, die eine Verringerung des berechneten Volumens der Gynäkomastie aufwiesen, Schmerzlinderung in der Brust) stimmten mit der primären Wirksamkeitsanalyse überein. Die Serumöstradiolkonzentrationen im 6. Behandlungsmonat waren in der ARIMIDEX-Gruppe um 15,4 % und in der Placebo-Gruppe um 4,5 % reduziert.
Nebenwirkungen, die von den Prüfärzten als behandlungsbedingt eingestuft wurden, traten bei 16,3 % der mit ARIMIDEX behandelten Patienten und 8,1 % der mit Placebo behandelten Patienten auf, wobei Akne (7 % ARIMIDEX und 2,7 % Placebo) und Kopfschmerzen (7 % ARIMIDEX 1 mg und 0 % Placebo); Alle anderen Nebenwirkungen zeigten kleine Unterschiede zwischen den Behandlungsgruppen. Ein Patient, der mit ARIMIDEX 1 mg behandelt wurde, brach die Studie wegen einer Hodenvergrößerung ab. Die durchschnittliche Veränderung des Hodenvolumens nach 6-monatiger Behandlung nach Abzug des Ausgangswerts betrug + 6,6 ± 7,9 cm³ bei den mit ARIMIDEX behandelten Patienten und + 5,2 ± 8,0 cm³ in der Placebogruppe.
McCune-Albright-Syndrom-Studie
Eine multizentrische, einarmige, offene Studie wurde mit 28 Mädchen mit McCune-Albright-Syndrom und fortschreitender vorzeitiger Pubertät im Alter von 2 bis
Bei fünf Patienten (18 %) traten Nebenwirkungen auf, die als möglicherweise mit ARIMIDEX zusammenhängend angesehen wurden. Diese waren Übelkeit, Akne, Schmerzen in einer Extremität, erhöhte Alanintransaminase und Aspartattransaminase und allergische Dermatitis.
Pharmakokinetik bei pädiatrischen Patienten
Nach mehrmaliger Gabe von 1 mg einmal täglich bei pädiatrischen Patienten betrug die mittlere Zeit bis zum Erreichen der maximalen Anastrozol-Konzentration 1 Stunde. Die mittleren (Bereichs-)Verteilungsparameter von Anastrozol bei pädiatrischen Patienten wurden durch ein CL/F von 1,54 l/h (0,77-4,53 l/h) und ein V/F von 98,4 l (50,7-330,0 l) beschrieben. Die terminale Eliminationshalbwertszeit betrug 46,8 h und war damit ähnlich wie bei postmenopausalen Frauen, die mit Anastrozol gegen Brustkrebs behandelt wurden. Basierend auf einer populationspharmakokinetischen Analyse war die Pharmakokinetik von Anastrozol bei Jungen mit pubertärer Gynäkomastie und Mädchen mit McCune-Albright-Syndrom ähnlich.
Geriatrische Verwendung
In den Studien 0030 und 0027 waren etwa 50 % der Patienten 65 Jahre oder älter. Patienten im Alter von ≥ 65 Jahren hatten unabhängig von der randomisierten Behandlung ein mäßig besseres Ansprechen des Tumors und eine mäßig bessere Zeit bis zur Tumorprogression als Patienten im Alter von
In der ATAC-Studie waren 45 % der Patienten 65 Jahre oder älter. Die Wirksamkeit von ARIMIDEX 1 mg im Vergleich zu Tamoxifen bei Patienten, die 65 Jahre oder älter waren (N = 1413 für ARIMIDEX und N = 1410 für Tamoxifen, die Hazard Ratio für das krankheitsfreie Überleben betrug 0,93 [95 %-KI: 0,80; 1,08]). weniger als die Wirksamkeit, die bei Patienten unter 65 Jahren beobachtet wurde (N = 1712 für ARIMIDEX und N = 1706 für Tamoxifen, die Hazard Ratio für das krankheitsfreie Überleben betrug 0,79 [95 % KI: 0,67; 0,94]).
Die Pharmakokinetik von Anastrozol wird durch das Alter nicht beeinflusst.
Nierenfunktionsstörung
Da Anastrozol nur zu etwa 10 % unverändert im Urin ausgeschieden wird, hat die Nierenfunktionsstörung keinen Einfluss auf die Gesamtkörperclearance. Eine Dosisanpassung bei Patienten mit eingeschränkter Nierenfunktion ist nicht erforderlich [siehe DOSIERUNG UND ANWENDUNG und KLINISCHE PHARMAKOLOGIE ].
Leberfunktionsstörung
Die Anastrozol-Plasmakonzentrationen bei den Probanden mit Leberzirrhose lagen in allen klinischen Studien im Bereich der Konzentrationen, die bei gesunden Probanden beobachtet wurden. Daher ist auch bei Patienten mit stabiler Leberzirrhose keine Dosisanpassung erforderlich. ARIMIDEX wurde bei Patienten mit schwerer Leberfunktionsstörung nicht untersucht [siehe DOSIERUNG UND ANWENDUNG und KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Es wurden klinische Studien mit ARIMIDEX durchgeführt, wobei bis zu 60 mg in einer Einzeldosis an gesunde männliche Probanden und bis zu 10 mg täglich an postmenopausale Frauen mit fortgeschrittenem Brustkrebs verabreicht wurden; diese Dosierungen wurden vertragen. Eine Einzeldosis von ARIMIDEX 1 mg, die zu lebensbedrohlichen Symptomen führt, wurde nicht ermittelt. Es gibt kein spezifisches Antidot für eine Überdosierung und die Behandlung muss symptomatisch sein. Berücksichtigen Sie bei der Behandlung einer Überdosierung, dass möglicherweise mehrere Wirkstoffe eingenommen wurden. Erbrechen kann ausgelöst werden, wenn der Patient wach ist. Eine Dialyse kann hilfreich sein, da ARIMIDEX nicht stark an Proteine gebunden ist. Eine allgemeine unterstützende Behandlung, einschließlich häufiger Überwachung der Vitalfunktionen und engmaschiger Beobachtung des Patienten, ist angezeigt.
KONTRAINDIKATIONEN
Schwangerschaft und prämenopausale Frauen
ARIMIDEX 1 mg kann den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird, und bietet prämenopausalen Frauen mit Brustkrebs keinen klinischen Nutzen. ARIMIDEX 1 mg ist kontraindiziert bei Frauen, die schwanger sind oder schwanger werden könnten. Es gibt keine angemessenen und gut kontrollierten Studien mit ARIMIDEX bei schwangeren Frauen. Wenn ARIMIDEX 1 mg während der Schwangerschaft angewendet wird oder wenn die Patientin während der Einnahme dieses Arzneimittels schwanger wird, sollte die Patientin über die potenzielle Gefahr für einen Fötus oder das potenzielle Risiko eines Schwangerschaftsverlusts informiert werden [siehe Abschnitt 4.4]. Verwendung in bestimmten Populationen ].
Überempfindlichkeit
ARIMIDEX 1 mg ist bei allen Patienten kontraindiziert, die eine Überempfindlichkeitsreaktion auf das Arzneimittel oder einen der sonstigen Bestandteile gezeigt haben. Beobachtete Reaktionen umfassen Anaphylaxie, Angioödem und Urtikaria [siehe NEBENWIRKUNGEN ]
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Das Wachstum vieler Brustkrebsarten wird durch Östrogene stimuliert oder aufrechterhalten.
Bei postmenopausalen Frauen stammen Östrogene hauptsächlich aus der Wirkung des Aromataseenzyms, das adrenale Androgene (hauptsächlich Androstendion und Testosteron) in Östron und Östradiol umwandelt. Die Unterdrückung der Östrogenbiosynthese in peripheren Geweben und im Krebsgewebe selbst kann daher durch eine gezielte Hemmung des Enzyms Aromatase erreicht werden.
Anastrozol ist ein selektiver nichtsteroidaler Aromatasehemmer. Es senkt die Estradiolkonzentrationen im Serum signifikant und hat keine nachweisbare Wirkung auf die Bildung von adrenalen Kortikosteroiden oder Aldosteron.
Pharmakodynamik
Wirkung auf Estradiol
Mittlere Serumkonzentrationen von Estradiol wurden in mehreren täglichen Dosierungsversuchen mit 0,5, 1, 3, 5 und 10 mg ARIMIDEX bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs bewertet. Bei allen Dosierungen wurde eine klinisch signifikante Unterdrückung des Serumöstradiols beobachtet. Dosen von 1 mg und höher führten zu einer Unterdrückung der mittleren Serumkonzentrationen von Estradiol auf die untere Nachweisgrenze (3,7 pmol/l). Die empfohlene Tagesdosis, ARIMIDEX 1 mg, reduzierte Estradiol um etwa 70 % innerhalb von 24 Stunden und um etwa 80 % nach 14 Tagen täglicher Dosierung. Die Unterdrückung von Estradiol im Serum wurde bis zu 6 Tage nach Beendigung der täglichen Einnahme von ARIMIDEX 1 mg aufrechterhalten.
Die Wirkung von ARIMIDEX 1 mg bei prämenopausalen Frauen mit frühem oder fortgeschrittenem Brustkrebs wurde nicht untersucht. Da die Aromatisierung von adrenalen Androgenen keine signifikante Östradiolquelle bei prämenopausalen Frauen ist, ist nicht zu erwarten, dass ARIMIDEX die Östradiolspiegel bei prämenopausalen Frauen senkt.
Wirkung auf Kortikosteroide
In Studien mit mehreren täglichen Dosierungen mit 3, 5 und 10 mg wurde die Selektivität von Anastrozol durch Untersuchung der Auswirkungen auf die Kortikosteroidsynthese bewertet. Bei allen Dosierungen beeinflusste Anastrozol die Cortisol- oder Aldosteronsekretion zu Studienbeginn oder als Reaktion auf ACTH nicht. Bei Anastrozol ist keine Glucocorticoid- oder Mineralocorticoid-Ersatztherapie erforderlich.
Andere endokrine Wirkungen
In mehreren täglichen Dosierungsversuchen mit 5 und 10 mg wurde das Schilddrüsen-stimulierende Hormon (TSH) gemessen; es gab keinen TSH-Anstieg während der Verabreichung von ARIMIDEX. ARIMIDEX besitzt bei Tieren keine direkte progestogene, androgene oder östrogene Aktivität, stört jedoch die zirkulierenden Spiegel von Progesteron, Androgenen und Östrogenen.
Pharmakokinetik
Absorption
Die Hemmung der Aromataseaktivität ist hauptsächlich auf Anastrozol, die Muttersubstanz, zurückzuführen. Die Resorption von Anastrozol erfolgt schnell und maximale Plasmakonzentrationen werden typischerweise innerhalb von 2 Stunden nach Einnahme im nüchternen Zustand erreicht. Studien mit radioaktiv markierten Arzneimitteln haben gezeigt, dass oral verabreichtes Anastrozol gut in den systemischen Kreislauf aufgenommen wird. Nahrung reduziert die Rate, aber nicht das Gesamtausmaß der Anastrozol-Resorption. Die mittlere Cmax von Anastrozol nahm um 16 % ab und die mediane Tmax verzögerte sich um 2 bis 5 Stunden, wenn Anastrozol 30 Minuten nach einer Mahlzeit verabreicht wurde. Die Pharmakokinetik von Anastrozol ist über den Dosisbereich von 1 bis 20 mg linear und ändert sich bei wiederholter Gabe nicht. Die Pharmakokinetik von Anastrozol war bei Patienten und gesunden Probanden ähnlich.
Verteilung
Steady-State-Plasmaspiegel sind etwa 3- bis 4-mal höher als die nach einer Einzeldosis ARIMIDEX beobachteten Spiegel. Die Plasmakonzentrationen nähern sich nach etwa 7 Tagen bei einmal täglicher Gabe den Steady-State-Spiegeln. Anastrozol ist im therapeutischen Bereich zu 40 % an Plasmaproteine gebunden.
Stoffwechsel
Der Metabolismus von Anastrozol erfolgt durch N-Dealkylierung, Hydroxylierung und Glucuronidierung. Drei Metaboliten von Anastrozol (Triazol, ein Glucuronid-Konjugat von Hydroxy-Anastrozol und ein Glucuronid-Konjugat von Anastrozol selbst) wurden in menschlichem Plasma und Urin identifiziert. Dem wichtigsten zirkulierenden Metaboliten von Anastrozol, Triazol, fehlt die pharmakologische Aktivität.
Anastrozol hemmte durch Cytochrom P450 1A2, 2C8/9 und 3A4 katalysierte Reaktionen in vitro mit Ki-Werten, die etwa 30-mal höher waren als die mittleren Steady-State-Cmax-Werte, die nach einer täglichen Dosis von 1 mg beobachtet wurden. Anastrozol hatte in vitro keine hemmende Wirkung auf durch Cytochrom P450 2A6 oder 2D6 katalysierte Reaktionen. Die Verabreichung einer Einzeldosis von 30 mg/kg oder mehrerer Dosen von 10 mg/kg Anastrozol an gesunde Probanden hatte keine Auswirkung auf die Clearance von Antipyrin oder die Wiederfindung von Antipyrin-Metaboliten im Urin.
Ausscheidung
85 % des radioaktiv markierten Anastrozols wurden in Fäkalien und Urin wiedergefunden. Der hepatische Metabolismus macht etwa 85 % der Elimination von Anastrozol aus. Die renale Elimination macht etwa 10 % der Gesamtclearance aus. Die mittlere Eliminationshalbwertszeit von Anastrozol beträgt 50 Stunden.
Einfluss von Geschlecht und Alter
Die Pharmakokinetik von Anastrozol wurde an postmenopausalen weiblichen Freiwilligen und Patientinnen mit Brustkrebs untersucht. Im Bereich von 80 Jahren wurden keine altersbedingten Auswirkungen beobachtet.
Auswirkung der Rasse
Die Serumspiegel von Östradiol und Östronsulfat waren bei japanischen und kaukasischen postmenopausalen Frauen, die 16 Tage lang täglich 1 mg Anastrozol erhielten, ähnlich. Die mittleren minimalen Anastrozol-Plasmakonzentrationen im Steady-State betrugen bei kaukasischen und japanischen postmenopausalen Frauen 25,7 bzw. 30,4 ng/ml.
Auswirkung einer Nierenfunktionsstörung
Die Pharmakokinetik von Anastrozol wurde bei Patienten mit eingeschränkter Nierenfunktion untersucht. Die renale Clearance von Anastrozol nahm proportional zur Kreatinin-Clearance ab und war bei Probanden mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance DOSIERUNG UND ANWENDUNG und Verwendung in bestimmten Populationen ].
Auswirkung einer Leberfunktionsstörung
Die Pharmakokinetik von Anastrozol wurde bei Patienten mit Leberzirrhose im Zusammenhang mit Alkoholmissbrauch untersucht. Die scheinbare orale Clearance (CL/F) von Anastrozol war bei Patienten mit stabiler Leberzirrhose etwa 30 % niedriger als bei Kontrollpersonen mit normaler Leberfunktion. Diese Plasmakonzentrationen lagen jedoch immer noch im Wertebereich, der bei gesunden Probanden beobachtet wurde. Die Auswirkungen einer schweren Leberfunktionsstörung wurden nicht untersucht. Bei stabiler Leberzirrhose ist keine Dosisanpassung erforderlich [vgl DOSIERUNG UND ANWENDUNG und Verwendung in bestimmten Bevölkerungsgruppen ].
Tiertoxikologie und/oder Pharmakologie
Reproduktionstoxikologie
Es wurde festgestellt, dass Anastrozol nach oraler Gabe von 0,1 mg/kg bei Ratten und Kaninchen (etwa das 1- bzw. 1,9-fache der empfohlenen Humandosis auf mg/m²-Basis) die Plazenta passiert. Studien an Ratten und Kaninchen mit Dosen von mindestens 0,1 bzw. 0,02 mg/kg/Tag (ca. 1 bzw. 1/3 der empfohlenen Humandosis auf mg/m²-Basis), verabreicht im Zeitraum von Die Organogenese zeigte, dass Anastrozol den Schwangerschaftsverlust erhöhte (erhöhter Prä- und/oder Postimplantationsverlust, erhöhte Resorption und verringerte Anzahl lebender Föten); Wirkungen waren bei Ratten dosisabhängig. Das Gewicht der Plazenta war bei Ratten bei Dosen von 0,1 mg/kg/Tag oder mehr signifikant erhöht.
Hinweise auf Fetotoxizität, einschließlich verzögerter fötaler Entwicklung (d. h. unvollständige Ossifikation und verringertes fötales Körpergewicht), wurden bei Ratten beobachtet, denen Dosen von 1 mg/kg/Tag verabreicht wurden (was zu Plasma-Anastrozol-Cssmax und -AUC 0-24-Stunden führte, die 19-fach und 9-mal höher als die entsprechenden Werte, die bei postmenopausalen Freiwilligen bei der empfohlenen Dosis gefunden wurden). Es gab keine Hinweise auf Teratogenität bei Ratten, denen Dosen von bis zu 1,0 mg/kg/Tag verabreicht wurden. Bei Kaninchen verursachte Anastrozol bei Dosen von mindestens 1,0 mg/kg/Tag (etwa dem 16-fachen der empfohlenen Humandosis auf mg/m²-Basis) eine Trächtigkeitsstörung; Es gab keine Hinweise auf Teratogenität bei Kaninchen, denen 0,2 mg/kg/Tag verabreicht wurde (etwa das Dreifache der empfohlenen Humandosis auf mg/m²-Basis).
Klinische Studien
Adjuvante Behandlung von Brustkrebs bei postmenopausalen Frauen
Eine multizentrische, doppelblinde Studie (ATAC) randomisierte 9.366 postmenopausale Frauen mit operablem Brustkrebs zu einer adjuvanten Behandlung mit ARIMIDEX 1 mg täglich, Tamoxifen 20 mg täglich oder einer Kombination der beiden Behandlungen für fünf Jahre oder bis zum Wiederauftreten der Krankheit.
Der primäre Endpunkt der Studie war das krankheitsfreie Überleben (dh die Zeit bis zum Auftreten eines entfernten oder lokalen Rezidivs oder eines kontralateralen Brustkrebses oder des Todes jeglicher Ursache). Zu den sekundären Endpunkten der Studie gehörten das krankheitsfreie Fernüberleben, die Inzidenz von kontralateralem Brustkrebs und das Gesamtüberleben. Bei einer medianen Nachbeobachtungszeit von 33 Monaten zeigte die Kombination von ARIMIDEX und Tamoxifen keinen Wirksamkeitsvorteil im Vergleich zu Tamoxifen bei allen Patienten sowie in der Hormonrezeptor-positiven Subpopulation. Dieser Behandlungsarm wurde aus der Studie ausgeschlossen. Basierend auf den klinischen und pharmakokinetischen Ergebnissen der ATAC-Studie sollte Tamoxifen nicht zusammen mit Anastrozol verabreicht werden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Demografische und andere Ausgangsmerkmale waren bei den drei Behandlungsgruppen ähnlich (siehe Tabelle 7).
Die Patienten in den beiden Monotherapie-Armen der ATAC-Studie wurden im Median 60 Monate (5 Jahre) behandelt und im Median 68 Monate nachbeobachtet. Das krankheitsfreie Überleben in der Intent-to-treat-Population war im ARIMIDEX-Arm im Vergleich zum Tamoxifen-Arm statistisch signifikant verbessert [Hazard Ratio (HR) = 0,87, 95 % KI: 0,78, 0,97, p = 0,0127]. In der Hormonrezeptor-positiven Subpopulation, die etwa 84 % der Studienpatienten ausmachte, war das krankheitsfreie Überleben im Arm mit ARIMIDEX 1 mg ebenfalls statistisch signifikant verbessert (HR = 0,83, 95 % KI: 0,73, 0,94, p = 0,0049). Tamoxifen-Arm.
Abbildung 1: Krankheitsfreies Überleben Kaplan-Meier-Überlebenskurve für alle Patienten, die in der ATAC-Studie (Intent-to-Treat) zu ARIMIDEX 1 mg oder Tamoxifen-Monotherapie randomisiert wurden 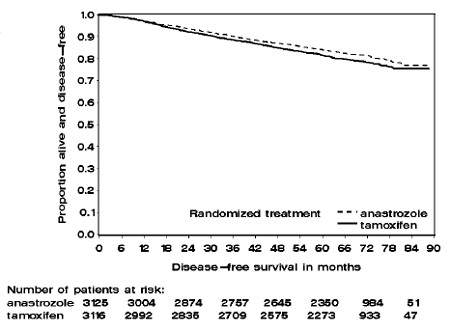
Abbildung 2: Krankheitsfreies Überleben einer Hormonrezeptor-positiven Subpopulation von Patienten, die in der ATAC-Studie zu ARIMIDEX- oder Tamoxifen-Monotherapie randomisiert wurden 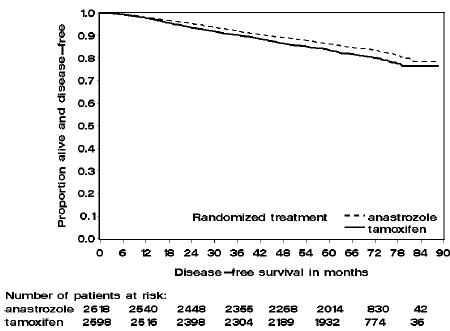
Die Überlebensdaten nach 68 Monaten Follow-up sind in Tabelle 9 dargestellt.
In der Gruppe der Patienten, die zuvor eine adjuvante Chemotherapie erhalten hatten (N = 698 für ARIMIDEX 1 mg und N = 647 für Tamoxifen), betrug die Hazard Ratio für das krankheitsfreie Überleben 0,91 (95 % KI: 0,73 bis 1,13) im ARIMIDEX-Arm im Vergleich zu der Tamoxifen-Arm.
Die Häufigkeit einzelner Ereignisse in der Intent-to-treat-Population und der Hormonrezeptor-positiven Subpopulation sind in Tabelle 8 beschrieben.
Eine Zusammenfassung der Wirksamkeitsergebnisse der Studie ist in Tabelle 9 aufgeführt.
10-Jahres-Median-Follow-up-Wirksamkeitsergebnisse aus der ATAC-Studie
In einer anschließenden Analyse der ATAC-Studie wurden Patienten in den beiden Monotherapie-Armen über einen medianen Zeitraum von 120 Monaten (10 Jahren) nachbeobachtet. Die Patienten erhielten die Studienbehandlung für einen Median von 60 Monaten (5 Jahren) (siehe Tabelle 10).
Abbildung 3: Krankheitsfreies Überleben Kaplan-Meier-Überlebenskurve für alle Patienten, die in der ATAC-Studie (Intent-to-Treat)a auf ARIMIDEX- oder Tamoxifen-Monotherapie randomisiert wurden 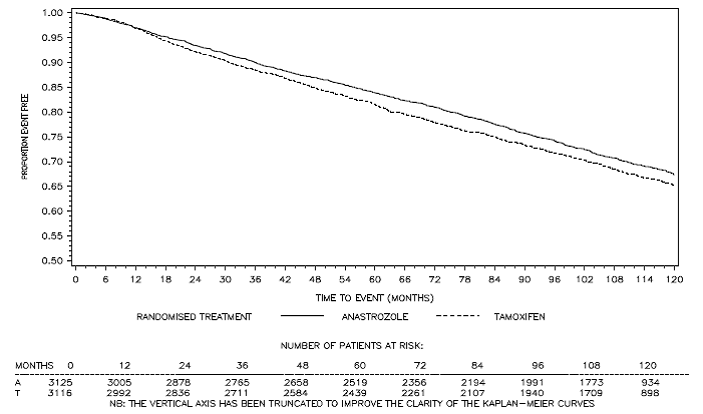
a Der Anteil der Patienten mit einer Nachbeobachtungszeit von 120 Monaten betrug 29,4 %.
Abbildung 4: Krankheitsfreies Überleben einer Hormonrezeptor-positiven Subpopulation von Patienten, die in der ATAC-Studie auf ARIMIDEX 1 mg oder Tamoxifen-Monotherapie randomisiert wurdenb 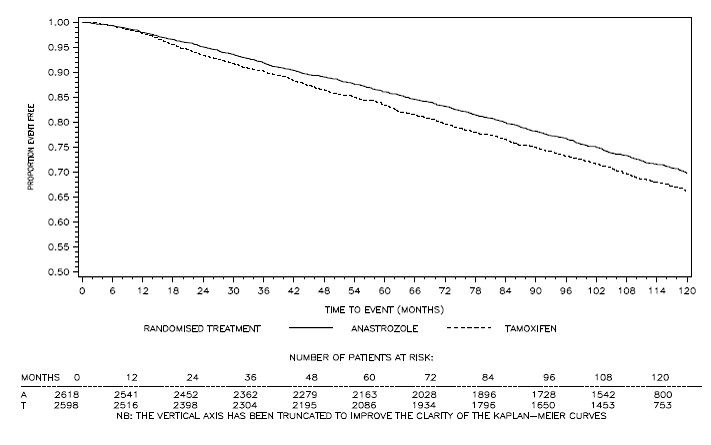
bDer Anteil der Patienten mit einer Nachbeobachtungszeit von 120 Monaten betrug 29,8 %.
First-Line-Therapie bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs
Zwei doppelblinde, kontrollierte klinische Studien mit ähnlichem Design (0030, eine nordamerikanische Studie und 0027, eine überwiegend europäische Studie) wurden durchgeführt, um die Wirksamkeit von ARIMIDEX 1 mg im Vergleich zu Tamoxifen als Erstlinientherapie für Hormonrezeptor-positive oder Hormonrezeptoren zu bewerten unbekannter lokal fortgeschrittener oder metastasierter Brustkrebs bei postmenopausalen Frauen. Insgesamt 1021 Patienten im Alter zwischen 30 und 92 Jahren wurden randomisiert, um eine Studienbehandlung zu erhalten. Die Patienten wurden randomisiert und erhielten einmal täglich 1 mg ARIMIDEX 1 mg oder einmal täglich 20 mg Tamoxifen. Die primären Endpunkte für beide Studien waren die Zeit bis zur Tumorprogression, die objektive Ansprechrate des Tumors und die Sicherheit.
Demografische und andere Charakteristika zu Studienbeginn, einschließlich Patienten mit messbarer und keiner messbaren Erkrankung, Patienten mit vorheriger adjuvanter Therapie, Ort der metastasierten Erkrankung und ethnische Herkunft, waren für die beiden Behandlungsgruppen in beiden Studien ähnlich. Die folgende Tabelle fasst den Hormonrezeptorstatus bei Eintritt für alle randomisierten Patienten in den Studien 0030 und 0027 zusammen.
Für die primären Endpunkte zeigte Studie 0030, dass ARIMIDEX einen statistisch signifikanten Vorteil gegenüber Tamoxifen (p=0,006) für die Zeit bis zur Tumorprogression hatte; Die objektiven Tumoransprechraten waren für ARIMIDEX 1 mg und Tamoxifen ähnlich. Studie 0027 zeigte, dass ARIMIDEX 1 mg und Tamoxifen ähnliche objektive Tumoransprechraten und Zeit bis zur Tumorprogression aufwiesen (siehe Tabelle 12 und Abbildungen 5 und 6).
Tabelle 12 unten fasst die Ergebnisse von Studie 0030 und Studie 0027 für die primären Wirksamkeitsendpunkte zusammen.
Abbildung 5: Kaplan-Meier-Wahrscheinlichkeit der Zeit bis zum Fortschreiten der Krankheit für alle randomisierten Patienten (Intent-to-treat) in Studie 0030 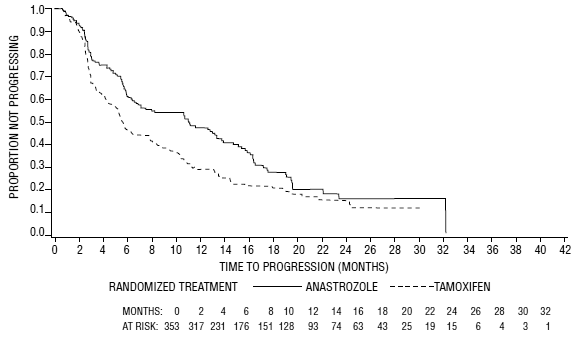
Abbildung 6: Kaplan-Meier-Wahrscheinlichkeit der Zeit bis zur Progression für alle randomisierten Patienten (Intent-to-treat) in Studie 0027 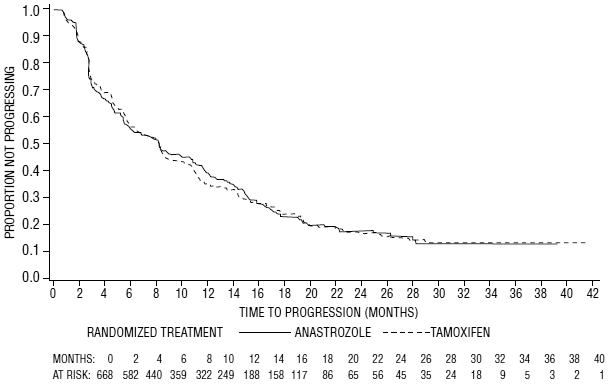
Die Ergebnisse der sekundären Endpunkte unterstützten die Ergebnisse der primären Wirksamkeitsendpunkte. In beiden Studien traten in den Behandlungsgruppen zu wenige Todesfälle auf, um Schlussfolgerungen zu Unterschieden im Gesamtüberleben ziehen zu können.
Zweitlinientherapie bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs, die nach einer Tamoxifen-Therapie eine Krankheitsprogression hatten
Anastrozol wurde in zwei kontrollierten klinischen Studien (0004, eine nordamerikanische Studie; 0005, eine überwiegend europäische Studie) bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs untersucht, bei denen es nach einer Tamoxifen-Therapie bei entweder fortgeschrittenem oder frühem Brustkrebs zu einer Krankheitsprogression kam. Einige der Patienten hatten auch zuvor eine zytotoxische Behandlung erhalten. Die meisten Patienten waren ER-positiv; ein kleinerer Anteil war ER-unbekannt oder ER-negativ; die ER-negativen Patienten kamen nur in Frage, wenn sie positiv auf Tamoxifen reagiert hatten. Geeignete Patienten mit messbarer und nicht messbarer Erkrankung wurden randomisiert und erhielten entweder eine tägliche Einzeldosis von 1 mg oder 10 mg ARIMIDEX oder 40 mg Megestrolacetat viermal täglich. Die Studien waren in Bezug auf ARIMIDEX doppelblind. Die Zeit bis zur Progression und die objektiven Ansprechraten (nur Patienten mit messbarer Erkrankung konnten als partielle Responder angesehen werden) waren die primären Wirksamkeitsvariablen. Objektive Ansprechraten wurden basierend auf den Kriterien der Union Internationale Contre le Cancer (UICC) berechnet. Die Rate der anhaltenden (mehr als 24 Wochen) stabilen Erkrankung, die Progressionsrate und das Überleben wurden ebenfalls berechnet.
Beide Studien umfassten über 375 Patienten; Demografische und andere Charakteristika zu Studienbeginn waren für die drei Behandlungsgruppen in jeder Studie ähnlich. Patienten in der 0005-Studie hatten besser auf eine vorangegangene Tamoxifen-Behandlung angesprochen. Von den aufgenommenen Patienten, die eine vorangegangene Tamoxifen-Therapie wegen fortgeschrittener Erkrankung hatten (58 % in Studie 0004; 57 % in Studie 0005), berichtete der primäre Prüfarzt, dass 18 % dieser Patienten in Studie 0004 und 42 % in Studie 0005 auf die Behandlung angesprochen hatten. In Studie 0004 waren 81 % der Patienten ER-positiv, 13 % waren ER-unbekannt und 6 % waren ER-negativ. In Studie 0005 waren 58 % der Patienten ER-positiv, 37 % waren ER-unbekannt und 5 % waren ER-negativ. In Studie 0004 hatten 62 % der Patienten eine messbare Erkrankung, verglichen mit 79 % in Studie 0005. Die Orte der metastasierten Erkrankung waren in den Behandlungsgruppen für jede Studie ähnlich. Im Durchschnitt hatten 40 % der Patienten Weichteilmetastasen; 60 % hatten Knochenmetastasen; und 40 % hatten viszerale (15 % Leber-) Metastasen.
Die Wirksamkeitsergebnisse der beiden Studien waren ähnlich, wie in Tabelle 13 dargestellt. In beiden Studien gab es keine signifikanten Unterschiede zwischen den Behandlungsarmen in Bezug auf einen der in der nachstehenden Tabelle aufgeführten Wirksamkeitsparameter.
Wenn die Daten aus den beiden kontrollierten Studien gepoolt werden, waren die objektiven Ansprechraten und medianen Zeiten bis zur Progression und zum Tod ähnlich für Patienten, die randomisiert ARIMIDEX 1 mg und Megestrolacetat erhielten. In diesen Daten gibt es keinen Hinweis darauf, dass ARIMIDEX 10 mg ARIMIDEX 1 mg überlegen ist.
INFORMATIONEN ZUM PATIENTEN
ARIMIDEX® A-rim-eh-dex (Anastrozol) Tabletten zur oralen Verabreichung
Was sind die wichtigsten Informationen, die ich über ARIMIDEX wissen sollte?
ARIMIDEX kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Holen Sie sich sofort medizinische Hilfe, wenn Sie während der Behandlung mit ARIMIDEX neue oder sich verschlimmernde Schmerzen in der Brust oder Kurzatmigkeit haben.
Was ist ARIMIDEX?
ARIMIDEX 1 mg ist ein verschreibungspflichtiges Arzneimittel, das bei Frauen nach der Menopause („die Veränderung des Lebens“) angewendet wird für:
ARIMIDEX 1 mg wirkt nicht bei Frauen mit Brustkrebs, die die Menopause noch nicht durchlaufen haben (prämenopausale Frauen).
Wer sollte ARIMIDEX nicht einnehmen?
Nehmen Sie ARIMIDEX 1 mg nicht ein, wenn Sie:
Was sollte ich meinem Arzt vor der Einnahme von ARIMIDEX 1 mg mitteilen?
Informieren Sie vor der Einnahme von ARIMIDEX Ihren Arzt, wenn Sie:
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und Kräuterergänzungen. Informieren Sie Ihren Arzt insbesondere, wenn Sie Folgendes einnehmen:
Informieren Sie sich über die Medikamente, die Sie einnehmen. Führen Sie eine Liste davon, um sie Ihrem Arzt und Apotheker zu zeigen, wenn Sie ein neues Arzneimittel erhalten.
Wie soll ich Arimidex einnehmen?
Wenn Sie zu viel ARIMIDEX 1 mg eingenommen haben, wenden Sie sich sofort an Ihren Arzt oder suchen Sie die Notaufnahme des nächstgelegenen Krankenhauses auf.
Welche Nebenwirkungen kann Arimidex haben?
ARIMIDEX kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Häufige Nebenwirkungen bei Frauen, die ARIMIDEX 1 mg einnehmen, sind:
ARIMIDEX kann auch ein Kitzeln, Kribbeln oder Taubheitsgefühl auf Ihrer Haut verursachen.
Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von ARIMIDEX. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist Arimidex 1 mg aufzubewahren?
Bewahren Sie ARIMIDEX und alle Arzneimittel außerhalb der Reichweite von Kindern auf.
Allgemeine Informationen zur sicheren und wirksamen Anwendung von ARIMIDEX.
Arzneimittel werden manchmal zu anderen als den in der Packungsbeilage aufgeführten Zwecken verschrieben. Nehmen Sie ARIMIDEX 1 mg nicht für einen Zustand ein, für den es nicht verschrieben wurde. Geben Sie ARIMIDEX nicht an andere Personen weiter, selbst wenn sie die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Wenn Sie weitere Informationen wünschen, sprechen Sie mit Ihrem Arzt. Sie können Ihren Apotheker oder Arzt um Informationen über ARIMIDEX bitten, die für Angehörige der Gesundheitsberufe bestimmt sind. Weitere Informationen erhalten Sie telefonisch unter 1-866-992-9276 oder unter www.ARIMIDEX.com.
Was sind die Inhaltsstoffe von ARIMIDEX?
Wirkstoff: Anastrozol
Inaktive Zutaten: Lactose, Magnesiumstearat, Hydroxypropylmethylcellulose, Polyethylenglykol, Povidon, Natriumstärkeglykolat und Titandioxid.