Risperdal 1mg, 2mg, 3mg, 4mg Risperidone Verwendung, Nebenwirkungen, Stärke und Dosierung. Preis in Online-Apotheke. Generika medikamente rezeptfrei.
Was ist Risperdal 1 mg und wie wird es angewendet?
Risperdal 4 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Schizophrenie, bipolarer Manie, bipolarer Störung und Reizbarkeit. Risperdal kann allein oder mit anderen Medikamenten verwendet werden.
Risperdal gehört zu einer Klasse von Arzneimitteln, die Antipsychotika, 2. Generation, Antimanika genannt werden.
Es ist nicht bekannt, ob Risperdal 2 mg bei Kindern unter 5 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Risperdal haben?
Risperdal kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Risperdal gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Risperdal. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt. Dies sind nicht alle möglichen Nebenwirkungen von Risperdal. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker. Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.WARNUNG
ERHÖHTE STERBLICHKEIT BEI ÄLTEREN PATIENTEN MIT PSYCHOSE IM ZUSAMMENHANG MIT DEMENZ
Ältere Patienten mit demenzbedingter Psychose, die mit Antipsychotika behandelt werden, haben ein erhöhtes Sterberisiko. RISPERDAL® (Risperidon) ist nicht für die Behandlung von Patienten mit demenzbedingter Psychose zugelassen. [Siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN]
BEZEICHNUNG
RISPERDAL® enthält Risperidon, ein atypisches Antipsychotikum, das zur chemischen Klasse der Benzisoxazol-Derivate gehört. Die chemische Bezeichnung lautet 3-[2-[4-(6-Fluor-1,2-benzisoxazol-3-yl)1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-2-methyl-4H- Pyrido[1,2-a]pyrimidin-4-on. Seine Molekularformel ist C23H27FN4O2 und sein Molekulargewicht beträgt 410,49. Die Strukturformel lautet:
Risperidon ist ein weißes bis leicht beiges Pulver. Es ist praktisch unlöslich in Wasser, frei löslich in Methylenchlorid und löslich in Methanol und 0,1 N HCl.
RISPERDAL® Tabletten sind zur oralen Verabreichung und in Stärken von 0,25 mg (dunkelgelb), 0,5 mg (rotbraun), 1 mg (weiß), 2 mg (orange), 3 mg (gelb) und 4 mg (grün) erhältlich . RISPERDAL®-Tabletten enthalten die folgenden Hilfsstoffe: kolloidales Siliciumdioxid, Hypromellose, Lactose, Magnesiumstearat, mikrokristalline Cellulose, Propylenglykol, Natriumlaurylsulfat und Stärke (Mais). Die 0,25-mg-, 0,5-mg-, 2-mg-, 3-mg- und 4-mg-Tabletten enthalten auch Talkum und Titandioxid. Die 0,25-mg-Tabletten enthalten gelbes Eisenoxid; die 0,5-mg-Tabletten enthalten rotes Eisenoxid; die 2-mg-Tabletten enthalten FD&C Yellow No. 6 Aluminium Lake; die 3-mg- und 4-mg-Tabletten enthalten D&C Yellow Nr. 10; die 4-mg-Tabletten enthalten FD&C Blue No. 2 Aluminium Lake.
RISPERDAL® ist auch als orale Lösung mit 1 mg/ml erhältlich. RISPERDAL® Lösung zum Einnehmen enthält die folgenden Hilfsstoffe: Weinsäure, Benzoesäure, Natriumhydroxid und gereinigtes Wasser.
RISPERDAL® M-TAB® Oral Disintegrating Tablets sind in Stärken von 0,5 mg (hellkoralle), 1 mg (hellkoralle), 2 mg (koralle), 3 mg (koralle) und 4 mg (koralle) erhältlich. RISPERDAL® M-TAB® oral zerfallende Tabletten enthalten die folgenden inaktiven Bestandteile: Amberlite®-Harz, Gelatine, Mannit, Glycin, Simethicon, Carbomer, Natriumhydroxid, Aspartam, rotes Eisenoxid und Pfefferminzöl. Darüber hinaus enthalten die 2 mg, 3 mg und 4 mg RISPERDAL® M-TAB® im Mund auflösende Tabletten Xanthangummi.
INDIKATIONEN
Schizophrenie
RISPERDAL® (Risperidon) ist zur Behandlung von Schizophrenie indiziert. Die Wirksamkeit wurde in 4 Kurzzeitstudien mit Erwachsenen, 2 Kurzzeitstudien mit Jugendlichen (im Alter von 13 bis 17 Jahren) und einer Langzeiterhaltungsstudie mit Erwachsenen nachgewiesen [siehe Klinische Studien ].
Bipolare Manie
Monotherapie
RISPERDAL® ist angezeigt für die Behandlung von akuten manischen oder gemischten Episoden im Zusammenhang mit einer Bipolar-I-Störung. Die Wirksamkeit wurde in 2 Kurzzeitstudien mit Erwachsenen und einer Kurzzeitstudie mit Kindern und Jugendlichen (im Alter von 10 bis 17 Jahren) nachgewiesen [siehe Klinische Studien ].
Begleitende Therapie
Die RISPERDAL®-Zusatztherapie mit Lithium oder Valproat ist angezeigt für die Behandlung von akuten manischen oder gemischten Episoden im Zusammenhang mit einer Bipolar-I-Störung. Die Wirksamkeit wurde in einer Kurzzeitstudie bei Erwachsenen nachgewiesen [siehe Klinische Studien ].
Reizbarkeit im Zusammenhang mit autistischer Störung
RISPERDAL® ist indiziert zur Behandlung von Reizbarkeit im Zusammenhang mit autistischen Störungen, einschließlich Symptomen von Aggression gegenüber anderen, vorsätzlicher Selbstverletzung, Wutausbrüchen und schnell wechselnden Stimmungen. Die Wirksamkeit wurde in 3 Kurzzeitstudien mit Kindern und Jugendlichen (im Alter von 5 bis 17 Jahren) nachgewiesen [siehe Klinische Studien ].
DOSIERUNG UND ANWENDUNG
Schwere Nieren- und Leberfunktionsstörung bei Erwachsenen: Wenden Sie eine niedrigere Anfangsdosis von 0,5 mg zweimal täglich an. Kann in Abständen von einer Woche oder länger auf Dosierungen über 1,5 mg zweimal täglich erhöht werden.
Schizophrenie
Erwachsene
Übliche Anfangsdosis
RISPERDAL® kann ein- oder zweimal täglich verabreicht werden. Die Anfangsdosis beträgt 2 mg pro Tag. Kann die Dosis in Abständen von 24 Stunden oder länger in Schritten von 1 bis 2 mg pro Tag je nach Verträglichkeit auf eine empfohlene Dosis von 4 bis 8 mg pro Tag erhöhen. Bei manchen Patienten kann eine langsamere Titration angemessen sein. Die Wirksamkeit wurde in einem Bereich von 4 mg bis 16 mg pro Tag nachgewiesen. Dosen über 6 mg pro Tag bei zweimal täglicher Gabe erwiesen sich jedoch nicht als wirksamer als niedrigere Dosen, waren mit mehr extrapyramidalen Symptomen und anderen Nebenwirkungen verbunden und werden im Allgemeinen nicht empfohlen. In einer einzelnen Studie, die eine einmal tägliche Dosierung unterstützte, waren die Wirksamkeitsergebnisse bei 8 mg im Allgemeinen stärker als bei 4 mg. Die Sicherheit von Dosen über 16 mg pro Tag wurde nicht in klinischen Studien untersucht [siehe Klinische Studien ].
Jugendliche
Die Anfangsdosis beträgt 0,5 mg einmal täglich, verabreicht als tägliche Einzeldosis morgens oder abends. Die Dosis kann je nach Verträglichkeit in Abständen von 24 Stunden oder länger in Schritten von 0,5 mg oder 1 mg pro Tag bis zu einer empfohlenen Dosis von 3 mg pro Tag angepasst werden. Obwohl die Wirksamkeit in Studien mit jugendlichen Patienten mit Schizophrenie bei Dosen zwischen 1 mg und 6 mg pro Tag nachgewiesen wurde, wurde kein zusätzlicher Nutzen über 3 mg pro Tag beobachtet, und höhere Dosen waren mit mehr unerwünschten Ereignissen verbunden. Dosen über 6 mg pro Tag wurden nicht untersucht.
Patienten mit anhaltender Somnolenz können von einer zweimal täglichen Verabreichung der halben Tagesdosis profitieren.
Erhaltungstherapie
Obwohl nicht bekannt ist, wie lange ein Patient mit Schizophrenie auf RISPERDAL® bleiben sollte, wurde die Wirksamkeit von RISPERDAL® 2 mg pro Tag bis 8 mg pro Tag bei der Verzögerung eines Rückfalls in einer kontrollierten Studie bei erwachsenen Patienten nachgewiesen, die mindestens seit klinisch stabil waren 4 Wochen und wurden dann über einen Zeitraum von 1 bis 2 Jahren nachbeobachtet [vgl Klinische Studien ]. Sowohl erwachsene als auch jugendliche Patienten, die akut ansprechen, sollten im Allgemeinen ihre wirksame Dosis über die akute Episode hinaus beibehalten. Die Patienten sollten regelmäßig neu untersucht werden, um die Notwendigkeit einer Erhaltungstherapie zu bestimmen.
Wiederaufnahme der Behandlung bei Patienten, die zuvor abgebrochen wurden
Obwohl es keine Daten gibt, die sich speziell mit der Wiederaufnahme der Behandlung befassen, wird empfohlen, dass nach einem Intervall ohne RISPERDAL® das anfängliche Titrationsschema befolgt wird.
Umstellung von anderen Antipsychotika
Es liegen keine systematisch gesammelten Daten vor, die sich speziell mit der Umstellung schizophrener Patienten von anderen Antipsychotika auf RISPERDAL® oder der Behandlung von Patienten mit gleichzeitigen Antipsychotika befassen.
Bipolare Manie
Übliche Dosis
Erwachsene
Die Anfangsdosis beträgt 2 mg bis 3 mg pro Tag. Die Dosis kann in Abständen von 24 Stunden oder länger in Schritten von 1 mg pro Tag angepasst werden. Der wirksame Dosisbereich beträgt 1 mg bis 6 mg pro Tag, wie in den placebokontrollierten Kurzzeitstudien untersucht. In diesen Studien wurde eine kurzzeitige (3 Wochen) antimanische Wirksamkeit in einem flexiblen Dosierungsbereich von 1 mg bis 6 mg pro Tag gezeigt [vgl Klinische Studien ]. RISPERDAL®-Dosen von mehr als 6 mg pro Tag wurden nicht untersucht.
Pädiatrie
Die Anfangsdosis beträgt 0,5 mg einmal täglich, verabreicht als tägliche Einzeldosis morgens oder abends. Die Dosis kann je nach Verträglichkeit in Abständen von 24 Stunden oder länger in Schritten von 0,5 mg oder 1 mg pro Tag bis zur empfohlenen Zieldosis von 1 mg bis 2,5 mg pro Tag angepasst werden. Obwohl die Wirksamkeit in Studien an pädiatrischen Patienten mit bipolarer Manie bei Dosen zwischen 0,5 mg und 6 mg pro Tag nachgewiesen wurde, wurde kein zusätzlicher Nutzen über 2,5 mg pro Tag beobachtet, und höhere Dosen waren mit mehr unerwünschten Ereignissen verbunden. Dosen über 6 mg pro Tag wurden nicht untersucht.
Patienten mit anhaltender Somnolenz können von einer zweimal täglichen Verabreichung der halben Tagesdosis profitieren.
Erhaltungstherapie
Es ist keine Evidenz aus kontrollierten Studien verfügbar, die einen Kliniker bei der längerfristigen Behandlung eines Patienten anleiten könnte, dem es während der Behandlung einer akuten manischen Episode mit RISPERDAL® besser geht. Während allgemein anerkannt wird, dass eine pharmakologische Behandlung über eine akute Reaktion hinaus bei Manie wünschenswert ist, sowohl zur Aufrechterhaltung des anfänglichen Ansprechens als auch zur Vorbeugung neuer manischer Episoden, gibt es keine systematisch erhobenen Daten, die die Anwendung von RISPERDAL® über einen solchen längeren Zeitraum unterstützen Behandlung (dh länger als 3 Wochen). Der Arzt, der sich für die Anwendung von RISPERDAL® über einen längeren Zeitraum entscheidet, sollte die langfristigen Risiken und Vorteile des Arzneimittels für den einzelnen Patienten regelmäßig neu bewerten.
Reizbarkeit im Zusammenhang mit autistischer Störung – Pädiatrie (Kinder und Jugendliche)
Die Dosierung von RISPERDAL® sollte entsprechend dem Ansprechen und der Verträglichkeit des Patienten individuell angepasst werden. Die gesamte Tagesdosis von RISPERDAL® kann einmal täglich verabreicht werden, oder die Hälfte der gesamten Tagesdosis kann zweimal täglich verabreicht werden.
Beginnen Sie bei Patienten mit einem Körpergewicht von weniger als 20 kg mit einer Dosis von 0,25 mg pro Tag. Beginnen Sie bei Patienten mit einem Körpergewicht von 20 kg oder mehr mit der Dosierung von 0,5 mg pro Tag. Nach mindestens vier Tagen kann die Dosis auf die empfohlene Dosis von 0,5 mg pro Tag für Patienten unter 20 kg und 1,0 mg pro Tag für Patienten über oder gleich 20 kg erhöht werden. Behalten Sie diese Dosis für mindestens 14 Tage bei. Bei Patienten, die kein ausreichendes klinisches Ansprechen erzielen, kann die Dosis in Abständen von mindestens 2 Wochen erhöht werden, in Schritten von 0,25 mg pro Tag für Patienten unter 20 kg oder in Schritten von 0,5 mg pro Tag für Patienten über oder gleich 20 kg. Der effektive Dosisbereich beträgt 0,5 mg bis 3 mg pro Tag. Für Kinder, die weniger als 15 kg wiegen, liegen keine Dosierungsdaten vor.
Sobald ein ausreichendes klinisches Ansprechen erreicht und aufrechterhalten wurde, sollte eine schrittweise Verringerung der Dosis erwogen werden, um ein optimales Gleichgewicht zwischen Wirksamkeit und Sicherheit zu erreichen. Der Arzt, der sich für die Anwendung von RISPERDAL® über einen längeren Zeitraum entscheidet, sollte die langfristigen Risiken und Vorteile des Arzneimittels für den einzelnen Patienten regelmäßig neu bewerten.
Patienten mit anhaltender Somnolenz können von einer einmal täglichen Dosis vor dem Schlafengehen oder einer zweimal täglichen Verabreichung der halben Tagesdosis oder einer Dosisreduktion profitieren.
Dosierung bei Patienten mit schwerer Nieren- oder Leberfunktionsstörung
Bei Patienten mit schwerer Nierenfunktionsstörung (CLcr Verwendung in bestimmten Populationen ].
Dosisanpassungen für spezifische Arzneimittelwechselwirkungen
Wenn RISPERDAL® zusammen mit Enzyminduktoren (z. B. Carbamazepin) verabreicht wird, sollte die Dosis von RISPERDAL® auf das Doppelte der üblichen Dosis des Patienten erhöht werden. Es kann erforderlich sein, die RISPERDAL®-Dosis zu verringern, wenn Enzyminduktoren wie Carbamazepin abgesetzt werden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ]. Eine ähnliche Wirkung kann bei gleichzeitiger Verabreichung von RISPERDAL® mit anderen Enzyminduktoren (z. B. Phenytoin, Rifampin und Phenobarbital) erwartet werden.
Wenn Fluoxetin oder Paroxetin zusammen mit RISPERDAL® verabreicht wird, sollte die Dosis von RISPERDAL® reduziert werden. Die RISPERDAL®-Dosis sollte bei Erwachsenen bei gleichzeitiger Anwendung mit diesen Arzneimitteln 8 mg pro Tag nicht überschreiten. Zu Beginn der Therapie sollte RISPERDAL® langsam titriert werden. Es kann erforderlich sein, die RISPERDAL®-Dosis zu erhöhen, wenn Enzyminhibitoren wie Fluoxetin oder Paroxetin abgesetzt werden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Verabreichung von RISPERDAL® Lösung zum Einnehmen
RISPERDAL® Lösung zum Einnehmen kann direkt aus der kalibrierten Pipette verabreicht oder vor der Verabreichung mit einem Getränk gemischt werden. RISPERDAL® Lösung zum Einnehmen ist mit den folgenden Getränken kompatibel: Wasser, Kaffee, Orangensaft und fettarme Milch; Es ist weder mit Cola noch mit Tee kompatibel.
Gebrauchsanweisung für RISPERDAL® M-TAB® oral zerfallende Tabletten
Tablet-Zugriff
RISPERDAL®M-TAB® Oral zerfallende Tabletten 0,5 mg, 1 mg und 2 mg
RISPERDAL® M-TAB® Oral Disintegrating Tablets 0,5 mg, 1 mg und 2 mg werden in Blisterpackungen mit jeweils 4 Tabletten geliefert.
Öffnen Sie die Blisterpackung erst, wenn Sie zur Verabreichung bereit sind. Trennen Sie zur Entnahme einer einzelnen Tablette eine der vier Blistereinheiten, indem Sie sie an den Perforationen auseinanderreißen. Biegen Sie die Ecke wie angegeben. Ziehen Sie die Folie zurück, um die Tablette freizulegen. Schieben Sie die Tablette NICHT durch die Folie, da dies die Tablette beschädigen könnte.
RISPERDAL®M-TAB® Orally Disintegrating Tablets 3 mg und 4 mg
RISPERDAL® M-TAB® Oral Disintegrating Tablets 3 mg und 4 mg werden in einem kindersicheren Beutel geliefert, der einen Blister mit jeweils 1 Tablette enthält.
Der kindersichere Beutel sollte an der Kerbe aufgerissen werden, um an die Blisterpackung zu gelangen. Öffnen Sie die Blisterpackung erst, wenn Sie zur Verabreichung bereit sind. Ziehen Sie die Folie von der Seite ab, um das Tablet freizulegen. Schieben Sie die Tablette NICHT durch die Folie, da dies die Tablette beschädigen könnte.
Tablet-Verwaltung
Entnehmen Sie die Tablette mit trockenen Händen aus der Blistereinheit und legen Sie sofort die gesamte RISPERDAL® M-TAB® im Mund zerfallende Tablette auf die Zunge. Die oral zerfallende RISPERDAL® MTAB® Tablette sollte sofort eingenommen werden, da die Tablette nach Entnahme aus der Blistereinheit nicht gelagert werden kann. RISPERDAL® M-TAB® Oral Disintegrating Tablets zerfallen innerhalb von Sekunden im Mund und können anschließend mit oder ohne Flüssigkeit geschluckt werden. Patienten sollten nicht versuchen, die Tablette zu teilen oder zu kauen.
WIE GELIEFERT
Darreichungsformen und Stärken
RISPERDAL® Tabletten sind in den folgenden Stärken und Farben erhältlich: 0,25 mg (dunkelgelb), 0,5 mg (rotbraun), 1 mg (weiß), 2 mg (orange), 3 mg (gelb) und 4 mg (grün). ). Alle sind kapselförmig und mit „JANSSEN“ auf der einen Seite und „Ris 0.25“, „Ris 0.5“, „R1“, „R2“, „R3“ oder „R4“ auf der anderen Seite entsprechend ihrer jeweiligen Größe bedruckt Stärken.
RISPERDAL® Lösung zum Einnehmen ist in einer Stärke von 1 mg/ml erhältlich.
RISPERDAL® M-TAB® Oral Disintegrating Tablets sind in den folgenden Stärken, Farben und Formen erhältlich: 0,5 mg (hellkoralle, rund), 1 mg (hellkoralle, quadratisch), 2 mg (koralle, quadratisch), 3 mg ( Koralle, rund) und 4 mg (Koralle, rund). Alle sind bikonvex und je nach Stärke auf einer Seite mit „R0.5“, „R1“, „R2“, „R3“ oder „R4“ geätzt.
RISPERDAL® (Risperidon) Tabletten
RISPERDAL® (Risperidon) Tabletten sind auf einer Seite mit „JANSSEN“ und je nach Stärke mit „Ris 0,25“, „Ris 0,5“, „R1“, „R2“, „R3“ oder „R4“ bedruckt.
0,25mg dunkelgelbe, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-301-04, Flaschen zu 500 NDC 50458-301-50 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-301-01.
0,5mg rotbraune, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-302-06, Flaschen zu 500 NDC 50458-302-50 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-302-01.
1mg weiße, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-300-06, Flaschen zu 500 NDC 50458-300-50 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-300-01.
2 mg Orange, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-320-06, Flaschen zu 500 NDC 50458-320-50 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-320-01.
3mg gelbe, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-330-06, Flaschen zu 500 NDC 50458-330-50 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-330-01.
4mg grüne, kapselförmige Tabletten: Flaschen mit 60 NDC 50458-350-06 und Blisterpackungen mit 100 Einzeldosen für Krankenhäuser NDC 50458-350-01.
RISPERDAL® (Risperidon) Lösung zum Einnehmen
RISPERDAL® (Risperidon) 1 mg/ml Lösung zum Einnehmen ( NDC 50458-305-03) wird in 30-ml-Flaschen mit einer kalibrierten (in Milligramm und Milliliter) Pipette geliefert. Das minimal kalibrierte Volumen beträgt 0,25 ml, während das maximal kalibrierte Volumen 3 ml beträgt.
RISPERDAL® M-TAB® (Risperidon) Im Mund zerfallende Tabletten
RISPERDAL® M-TAB® (Risperidon) Im Mund zerfallende Tabletten sind je nach Stärke einseitig mit „R0,5“, „R1“, „R2“, „R3“ oder „R4“ geätzt. RISPERDAL® MTAB® Oral Disintegrating Tablets 0,5 mg, 1 mg und 2 mg sind in Blisterpackungen mit 4 (2 x 2) Tabletten verpackt. Oral Disintegrating Tablets 3 mg und 4 mg sind in einem kindergesicherten Beutel verpackt, der einen Blister mit 1 Tablette enthält.
0,5mg helle Koralle, runde, bikonvexe Tabletten: 7 Blisterpackungen (je 4 Tabletten) pro Packung, NDC 50458-395-28 und Langzeitpflege-Blisterpackung mit 30 Tabletten NDC 50458-395-30.
1mg hellkoralle, quadratisch, bikonvexe Tabletten: 7 Blisterpackungen (je 4 Tabletten) pro Packung, NDC 50458-315-28 und Langzeitpflege-Blisterpackung mit 30 Tabletten NDC 50458-315-30.
2 mg Koralle, quadratisch, bikonvexe Tabletten: 7 Blisterpackungen (je 4 Tabletten) pro Packung, NDC 50458-325-28.
3mg Koralle, runde, bikonvexe Tabletten: 28 Blister pro Packung, NDC 50458-335-28.
4mg Koralle, runde, bikonvexe Tabletten: 28 Blister pro Packung, NDC 50458-355-28.
Lagerung und Handhabung
RISPERDAL® Tabletten sollten bei einer kontrollierten Raumtemperatur von 15°-25°C (59°-77°F) gelagert werden. Vor Licht und Feuchtigkeit schützen.
RISPERDAL® 1 mg/ml Lösung zum Einnehmen sollte bei einer kontrollierten Raumtemperatur von 15°25°C (59°-77°F) gelagert werden. Vor Licht und Frost schützen.
RISPERDAL® M-TAB® oral zerfallende Tabletten sollten bei einer kontrollierten Raumtemperatur von 15°-25°C (59°-77°F) gelagert werden.
Außerhalb der Reichweite von Kindern aufbewahren.
RISPERDAL® Tabletten Wirkstoff wird in Irland hergestellt Fertigprodukt wird hergestellt von: Janssen Ortho, LLC Gurabo, Puerto Rico 00778. RISPERDAL® Lösung zum Einnehmen Fertigprodukt wird hergestellt von: Janssen Pharmaceutica NV Beerse, Belgien. RISPERDAL® M-TAB® Orally Disintegrating Tablets Wirkstoff wird in Irland hergestellt. Fertigprodukt wird hergestellt von: Janssen Ortho, LLC Gurabo, Puerto Rico 00778. RISPERDAL® Tablets, RISPERDAL® M-TAB® Orally Disintegrating Tablets und RISPERDAL® Oral Solution werden hergestellt für: Janssen Pharmaceuticals, Inc. Titusville, NJ 08560. Überarbeitet: März 2016
NEBENWIRKUNGEN
Folgendes wird in anderen Abschnitten der Kennzeichnung ausführlicher behandelt:
Die häufigsten Nebenwirkungen in klinischen Studien (> 5 % und zweimal Placebo) waren Parkinsonismus, Akathisie, Dystonie, Tremor, Sedierung, Schwindel, Angstzustände, verschwommenes Sehen, Übelkeit, Erbrechen, Oberbauchschmerzen, Magenbeschwerden, Dyspepsie, Durchfall, Speichelfluss Hypersekretion, Verstopfung, Mundtrockenheit, gesteigerter Appetit, Gewichtszunahme, Müdigkeit, Hautausschlag, verstopfte Nase, Infektion der oberen Atemwege, Nasopharyngitis und pharyngolaryngeale Schmerzen.
Die häufigsten Nebenwirkungen, die mit dem Abbruch von klinischen Studien einhergingen (was bei > 1 % der Erwachsenen und/oder > 2 % der Kinder zum Abbruch führte), waren Übelkeit, Somnolenz, Sedierung, Erbrechen, Schwindel und Akathisie [siehe Abbrüche aufgrund von Nebenwirkungen ].
Die in diesem Abschnitt beschriebenen Daten stammen aus einer klinischen Studiendatenbank, die aus 9803 erwachsenen und pädiatrischen Patienten besteht, die einer oder mehreren Dosen RISPERDAL® zur Behandlung von Schizophrenie, bipolarer Manie, autistischer Störung und anderen psychiatrischen Störungen bei pädiatrischen und älteren Patienten ausgesetzt wurden mit Demenz. Von diesen 9803 Patienten waren 2687 Patienten, die RISPERDAL® erhielten, während sie an doppelblinden, placebokontrollierten Studien teilnahmen. Die Bedingungen und Dauer der Behandlung mit RISPERDAL® waren sehr unterschiedlich und umfassten (in überlappenden Kategorien) doppelblinde, fixe und flexible, placebo- oder aktivkontrollierte Studien und offene Studienphasen, stationär und ambulant, und kurz -langfristige (bis zu 12 Wochen) und längerfristige (bis zu 3 Jahre) Expositionen. Die Sicherheit wurde bewertet, indem unerwünschte Ereignisse gesammelt und körperliche Untersuchungen, Vitalfunktionen, Körpergewichte, Laboranalysen und EKGs durchgeführt wurden.
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Häufig beobachtete Nebenwirkungen in doppelblinden, Placebo-kontrollierten klinischen Studien – Schizophrenie
Erwachsene Patienten mit Schizophrenie
Tabelle 8 listet die Nebenwirkungen auf, die bei 2 % oder mehr der mit RISPERDAL® behandelten erwachsenen Patienten mit Schizophrenie in drei 4- bis 8-wöchigen, doppelblinden, placebokontrollierten Studien berichtet wurden.
Pädiatrische Patienten mit Schizophrenie
Tabelle 9 führt die Nebenwirkungen auf, die bei 5 % oder mehr der mit RISPERDAL® behandelten pädiatrischen Patienten mit Schizophrenie in einer 6-wöchigen, doppelblinden, placebokontrollierten Studie berichtet wurden.
Häufig beobachtete Nebenwirkungen in doppelblinden, Placebo-kontrollierten klinischen Studien – Bipolare Manie
Erwachsene Patienten mit bipolarer Manie
Tabelle 10 listet die Nebenwirkungen auf, die bei 2 % oder mehr der mit RISPERDAL® behandelten erwachsenen Patienten mit bipolarer Manie in vier 3-wöchigen, doppelblinden, placebokontrollierten Monotherapiestudien berichtet wurden.
Tabelle 11 listet die Nebenwirkungen auf, die bei 2 % oder mehr der mit RISPERDAL® behandelten erwachsenen Patienten mit bipolarer Manie in zwei 3-wöchigen, doppelblinden, placebokontrollierten adjuvanten Therapiestudien berichtet wurden.
Pädiatrische Patienten mit bipolarer Manie
Tabelle 12 führt die Nebenwirkungen auf, die bei mindestens 5 % der mit RISPERDAL® behandelten pädiatrischen Patienten mit bipolarer Manie in einer 3-wöchigen, doppelblinden, placebokontrollierten Studie berichtet wurden.
Häufig beobachtete Nebenwirkungen in doppelblinden, Placebo-kontrollierten klinischen Studien - Autistische Störung
Tabelle 13 führt die Nebenwirkungen auf, die bei 5 % oder mehr der mit RISPERDAL® behandelten pädiatrischen Patienten berichtet wurden, die in zwei 8-wöchigen, doppelblinden, placebokontrollierten Studien und einer 6-wöchigen, doppelblinden, Placebo-Studie wegen Reizbarkeit im Zusammenhang mit autistischer Störung behandelt wurden -kontrollierte Studie.
Andere Nebenwirkungen, die während der klinischen Studienbewertung von Risperidon beobachtet wurden
Die folgenden zusätzlichen Nebenwirkungen traten in allen placebokontrollierten, aktiv kontrollierten und unverblindeten Studien mit RISPERDAL® bei Erwachsenen und pädiatrischen Patienten auf.
Erkrankungen des Blut- und Lymphsystems: Anämie, Granulozytopenie, Neutropenie
Herzerkrankungen: Sinusbradykardie, Sinustachykardie, AV-Block I. Grades, Schenkelblock links, Schenkelblock rechts, AV-Block
Erkrankungen des Ohrs und des Labyrinths: Ohrenschmerzen, Tinnitus
Endokrine Störungen: Hyperprolaktinämie
Augenerkrankungen: Augenhyperämie, Augenausfluss, Konjunktivitis, Augenrollen, Augenlidödem, Augenschwellung, Verkrustung des Augenlidrandes, trockenes Auge, vermehrter Tränenfluss, Photophobie, Glaukom, verminderte Sehschärfe
Gastrointestinale Störungen: Dysphagie, Fäkalom, Stuhlinkontinenz, Gastritis, Lippenschwellung, Cheilitis, Aptyalismus
Allgemeine Erkrankungen: peripheres Ödem, Durst, Gangstörung, grippeähnliche Erkrankung, Lochfraß, Ödem, Schüttelfrost, Trägheit, Unwohlsein, Brustbeschwerden, Gesichtsödem, Unwohlsein, generalisiertes Ödem, Arzneimittelentzugssyndrom, peripheres Kältegefühl, anormales Gefühl
Erkrankungen des Immunsystems: Arzneimittelüberempfindlichkeit
Infektionen und Schädlinge: Lungenentzündung, Grippe, Ohrinfektion, virale Infektion, Pharyngitis, Tonsillitis, Bronchitis, Augeninfektion, lokalisierte Infektion, Zystitis, Zellulitis, Mittelohrentzündung, Onychomykose, Akarodermatitis, Bronchopneumonie, Atemwegsinfektion, Tracheobronchitis, chronische Mittelohrentzündung
Untersuchungen: Körpertemperatur erhöht, Prolaktin im Blut erhöht, Alanin-Aminotransferase erhöht, Elektrokardiogramm abnormal, Eosinophilenzahl erhöht, Anzahl weißer Blutkörperchen erniedrigt, Blutzucker erhöht, Hämoglobin erniedrigt, Hämatokrit erniedrigt, Körpertemperatur erniedrigt, Blutdruck erniedrigt, Transaminasen erhöht
Stoffwechsel- und Ernährungsstörungen: verminderter Appetit, Polydipsie, Anorexie
Erkrankungen des Bewegungsapparates und des Bindegewebes: Gelenksteifheit, Gelenkschwellung, muskuloskelettale Brustschmerzen, anormale Körperhaltung, Myalgie, Nackenschmerzen, Muskelschwäche, Rhabdomyolyse
Erkrankungen des Nervensystems: Gleichgewichtsstörung, Aufmerksamkeitsstörung, Dysarthrie, Reaktionslosigkeit auf Reize, Bewusstseinsstörung, Bewegungsstörung, transitorische ischämische Attacke, abnorme Koordination, Schlaganfall, Sprachstörung, Synkope, Bewusstseinsverlust, Hypästhesie, tardive Dyskinesie, Dyskinesie, zerebrale Ischämie, zerebrovaskuläre Störung, malignes neuroleptisches Syndrom, diabetisches Koma, Kopftötung
Psychische Störungen: Agitiertheit, abgestumpfter Affekt, Verwirrtheitszustand, mittlere Schlaflosigkeit, Nervosität, Schlafstörung, Lustlosigkeit, verminderte Libido und Anorgasmie
Nieren- und Harnwegserkrankungen: Enuresis, Dysurie, Pollakisurie, Harninkontinenz
Störungen des Fortpflanzungssystems und der Brust: Unregelmäßige Menstruation, Amenorrhoe, Gynäkomastie, Galaktorrhoe, vaginaler Ausfluss, Menstruationsstörung, erektile Dysfunktion, retrograde Ejakulation, Ejakulationsstörung, sexuelle Dysfunktion, Brustvergrößerung
Erkrankungen der Atemwege, des Brustraums und des Mediastinums: Keuchen, Lungenentzündung, Verstopfung der Nasennebenhöhlen, Dysphonie, produktiver Husten, verstopfte Lunge, verstopfte Atemwege, Rasseln, Atemstörung, Hyperventilation, Nasenödem
Erkrankungen der Haut und des Unterhautgewebes: Erythem, Hautverfärbung, Hautläsion, Juckreiz, Hauterkrankung, erythematöser Ausschlag, papulöser Ausschlag, generalisierter Ausschlag, makulopapulöser Ausschlag, Akne, Hyperkeratose, seborrhoische Dermatitis
Gefäßerkrankungen: Hypotonie, Hitzewallungen
Zusätzliche Nebenwirkungen, die mit RISPERDAL® CONSTA® berichtet wurden
Die folgende Liste enthält weitere Nebenwirkungen, die während der Beurteilung vor der Markteinführung von RISPERDAL® CONSTA® gemeldet wurden, unabhängig von der Häufigkeit ihres Auftretens:
Herzerkrankungen: Bradykardie
Erkrankungen des Ohrs und des Labyrinths: Schwindel
Augenerkrankungen: Blepharospasmus
Gastrointestinale Störungen: Zahnschmerzen, Zungenkrämpfe
Allgemeine Erkrankungen und Bedingungen am Verabreichungsort: Schmerzen
Infektionen und Schädlinge: Infektion der unteren Atemwege, Infektion, Gastroenteritis, subkutaner Abszess
Verletzung und Vergiftung: Herbst
Untersuchungen: Gewichtsabnahme, Gamma-Glutamyltransferase erhöht, Leberenzyme erhöht
Erkrankungen des Bewegungsapparates, des Bindegewebes und der Knochen: Gesäßschmerzen
Erkrankungen des Nervensystems: Krämpfe, Parästhesien
Psychische Störungen: Depression
Erkrankungen der Haut und des Unterhautgewebes: Ekzem
Gefäßerkrankungen: Hypertonie
Abbrüche aufgrund von Nebenwirkungen
Schizophrenie - Erwachsene
Etwa 7 % (39/564) der mit RISPERDAL® behandelten Patienten in doppelblinden, placebokontrollierten Studien brachen die Behandlung aufgrund einer Nebenwirkung ab, verglichen mit 4 % (10/225) der Placebo-Gruppe. Die Nebenwirkungen im Zusammenhang mit dem Absetzen bei 2 oder mehr mit RISPERDAL® behandelten Patienten waren:
In einer doppelblinden, placebo- und aktivkontrollierten Studie wurde die Behandlung wegen extrapyramidaler Symptome (einschließlich Parkinsonismus, Akathisie, Dystonie und tardive Dyskinesie) bei 1 % der mit Placebo behandelten Patienten und bei 3,4 % der mit aktiver Kontrolle behandelten Patienten abgebrochen.
Schizophrenie - Pädiatrie
Etwa 7 % (7/106) der mit RISPERDAL® behandelten Patienten brachen die Behandlung aufgrund einer Nebenwirkung in einer doppelblinden, placebokontrollierten Studie ab, verglichen mit 4 % (2/54) der mit Placebo behandelten Patienten. Die mit dem Absetzen verbundenen Nebenwirkungen waren bei mindestens einem mit RISPERDAL® behandelten Patienten Schwindel (2 %), Somnolenz (1 %), Sedierung (1 %), Lethargie (1 %), Angst (1 %), Gleichgewichtsstörungen (1 %), Hypotonie (1 %) und Herzklopfen (1 %).
Bipolare Manie - Erwachsene
In doppelblinden, placebokontrollierten Studien mit RISPERDAL® als Monotherapie brachen etwa 6 % (25/448) der mit RISPERDAL® behandelten Patienten die Behandlung aufgrund eines unerwünschten Ereignisses ab, verglichen mit etwa 5 % (19/424) der Placebo- behandelte Patienten. Die Nebenwirkungen im Zusammenhang mit dem Absetzen bei mit RISPERDAL® behandelten Patienten waren:
Bipolare Manie - Pädiatrie
In einer doppelblinden, placebokontrollierten Studie brachen 12 % (13/111) der mit RISPERDAL® behandelten Patienten die Behandlung aufgrund einer Nebenwirkung ab, verglichen mit 7 % (4/58) der mit Placebo behandelten Patienten. Die Nebenwirkungen im Zusammenhang mit dem Absetzen bei mehr als einem mit RISPERDAL® behandelten pädiatrischen Patienten waren Übelkeit (3 %), Somnolenz (2 %), Sedierung (2 %) und Erbrechen (2 %).
Autistische Störung - Pädiatrie
In den beiden 8-wöchigen placebokontrollierten Studien an pädiatrischen Patienten, die wegen Reizbarkeit in Verbindung mit autistischer Störung behandelt wurden (n = 156), brach ein mit RISPERDAL® behandelter Patient aufgrund einer Nebenwirkung (Parkinsonismus) und ein mit Placebo behandelter Patient ab aufgrund eines unerwünschten Ereignisses.
Dosisabhängigkeit von Nebenwirkungen in klinischen Studien
Extrapyramidale Symptome
Daten aus zwei Studien mit fester Dosierung bei Erwachsenen mit Schizophrenie lieferten Hinweise auf eine Dosisabhängigkeit extrapyramidaler Symptome im Zusammenhang mit der Behandlung mit RISPERDAL®.
Zwei Methoden wurden verwendet, um extrapyramidale Symptome (EPS) in einer 8-wöchigen Studie zu messen, in der 4 Fixdosen von RISPERDAL® (2, 6, 10 und 16 mg/Tag) verglichen wurden, einschließlich (1) eines Parkinson-Scores (mittlere Veränderung gegenüber dem Ausgangswert ) aus der Extrapyramidal Symptom Rating Scale und (2) Inzidenz spontaner EPS-Beschwerden:
Ähnliche Methoden wurden verwendet, um extrapyramidale Symptome (EPS) in einer 8-wöchigen Studie zu messen, in der 5 Fixdosen von RISPERDAL® (1, 4, 8, 12 und 16 mg/Tag) verglichen wurden:
Dystonie
Klasseneffekt: Bei empfindlichen Personen können in den ersten Tagen der Behandlung Symptome einer Dystonie, verlängerte abnorme Kontraktionen von Muskelgruppen, auftreten. Zu den dystonischen Symptomen gehören: Krämpfe der Nackenmuskulatur, die sich manchmal zu einem Engegefühl im Hals entwickeln, Schluckbeschwerden, Atembeschwerden und/oder Heraustreten der Zunge. Während diese Symptome bei niedrigen Dosen auftreten können, treten sie häufiger und mit größerer Schwere bei hochpotenten und höheren Dosen von Antipsychotika der ersten Generation auf. Ein erhöhtes Risiko einer akuten Dystonie wird bei Männern und jüngeren Altersgruppen beobachtet.
Andere Nebenwirkungen
Daten zu unerwünschten Ereignissen, die durch eine Checkliste für Nebenwirkungen aus einer großen Studie erhoben wurden, in der 5 Fixdosen von RISPERDAL® (1, 4, 8, 12 und 16 mg/Tag) verglichen wurden, wurden auf die Dosisabhängigkeit von unerwünschten Ereignissen untersucht. Ein Cochran-Armitage-Test für den Trend in diesen Daten zeigte einen positiven Trend (p
Änderungen des Körpergewichts
Gewichtszunahme wurde in kontrollierten Kurzzeitstudien und unkontrollierten Langzeitstudien bei erwachsenen und pädiatrischen Patienten beobachtet [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , und Verwendung in bestimmten Bevölkerungsgruppen ].
Änderungen der EKG-Parameter
Vergleiche zwischen den Gruppen gepoolter, placebokontrollierter Studien mit Erwachsenen ergaben keine statistisch signifikanten Unterschiede zwischen Risperidon und Placebo in Bezug auf die mittleren Veränderungen der EKG-Parameter, einschließlich QT-, QTc- und PR-Intervalle, und der Herzfrequenz gegenüber dem Ausgangswert. Als alle RISPERDAL®-Dosen aus randomisierten kontrollierten Studien in mehreren Indikationen gepoolt wurden, gab es einen mittleren Anstieg der Herzfrequenz von 1 Schlag pro Minute im Vergleich zu keiner Veränderung bei Placebo-Patienten. In Kurzzeitstudien zur Schizophrenie waren höhere Dosen von Risperidon (8-16 mg/Tag) im Vergleich zu Placebo (4-6 Schläge pro Minute) mit einem stärkeren mittleren Anstieg der Herzfrequenz verbunden. In gepoolten Placebo-kontrollierten Studien zur akuten Manie bei Erwachsenen gab es kleine Abnahmen der mittleren Herzfrequenz, ähnlich in allen Behandlungsgruppen.
In den beiden placebokontrollierten Studien bei Kindern und Jugendlichen mit autistischer Störung (im Alter von 5 bis 16 Jahren) waren die mittleren Veränderungen der Herzfrequenz ein Anstieg von 8,4 Schlägen pro Minute in den RISPERDAL®-Gruppen und 6,5 Schlägen pro Minute in der Placebogruppe. Es gab keine anderen bemerkenswerten EKG-Veränderungen.
In einer placebokontrollierten Studie zur akuten Manie bei Kindern und Jugendlichen (im Alter von 10 – 17 Jahren) gab es keine signifikanten Veränderungen der EKG-Parameter, abgesehen von der Wirkung von RISPERDAL® zur vorübergehenden Erhöhung der Pulsfrequenz (
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von Risperidon nach der Zulassung festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen. Diese Nebenwirkungen umfassen: Alopezie, anaphylaktische Reaktion, Angioödem, Vorhofflimmern, Herz-Lungen-Stillstand, diabetische Ketoazidose bei Patienten mit gestörtem Glukosestoffwechsel, Dysgeusie, Hypoglykämie, Hypothermie, Ileus, unangemessene ADH-Sekretion, Darmverschluss, Gelbsucht, Manie, Pankreatitis, Hypophyse Adenom, vorzeitige Pubertät, Lungenembolie, QT-Verlängerung, Schlafapnoe-Syndrom, plötzlicher Tod, Thrombozytopenie, thrombotisch-thrombozytopenische Purpura, Harnverhalt und Wasservergiftung.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Pharmakokinetische Wechselwirkungen
Die Dosis von RISPERDAL® sollte angepasst werden, wenn es in Kombination mit CYP2D6-Enzyminhibitoren (z. B. Fluoxetin und Paroxetin) und Enzyminduktoren (z. B. Carbamazepin) angewendet wird [siehe Tabelle 18 und DOSIERUNG UND ANWENDUNG ]. Eine Dosisanpassung wird für RISPERDAL® bei gleichzeitiger Anwendung mit Ranitidin, Cimetidin, Amitriptylin oder Erythromycin nicht empfohlen [siehe Tabelle 18].
Wirkung von Risperidon auf andere Medikamente
Lithium
Wiederholte orale Dosen von RISPERDAL® (3 mg zweimal täglich) hatten keinen Einfluss auf die Exposition (AUC) oder die maximalen Plasmakonzentrationen (Cmax) von Lithium (n=13). Eine Dosisanpassung für Lithium wird nicht empfohlen.
Valproat
Wiederholte orale Dosen von RISPERDAL® (4 mg einmal täglich) hatten im Vergleich zu Placebo (n=21) keinen Einfluss auf die Prädosis oder die durchschnittlichen Plasmakonzentrationen und die Exposition (AUC) von Valproat (1000 mg/Tag in drei aufgeteilten Dosen). Nach gleichzeitiger Gabe von RISPERDAL® kam es jedoch zu einem 20%igen Anstieg der Valproat-Spitzenplasmakonzentration (Cmax). Eine Dosisanpassung für Valproat wird nicht empfohlen.
Digoxin
RISPERDAL® (0,25 mg zweimal täglich) zeigte keine klinisch relevante Wirkung auf die Pharmakokinetik von Digoxin. Eine Dosisanpassung für Digoxin wird nicht empfohlen.
Pharmakodynamische Wechselwirkungen
Zentral wirkende Drogen und Alkohol
Angesichts der primären ZNS-Wirkungen von Risperidon ist Vorsicht geboten, wenn RISPERDAL® in Kombination mit anderen zentral wirksamen Arzneimitteln und Alkohol eingenommen wird.
Medikamente mit blutdrucksenkender Wirkung
Aufgrund seines Potenzials zur Induktion von Hypotonie kann RISPERDAL® die blutdrucksenkende Wirkung anderer Therapeutika mit diesem Potenzial verstärken.
Levodopa und Dopamin-Agonisten
RISPERDAL® kann die Wirkung von Levodopa und Dopaminagonisten antagonisieren.
Clozapin
Die chronische Gabe von Clozapin zusammen mit RISPERDAL® kann die Clearance von Risperidon verringern.
Drogenmissbrauch und -abhängigkeit
Kontrollierte Substanz
RISPERDAL® (Risperidon) ist keine kontrollierte Substanz.
Missbrauch
RISPERDAL® wurde nicht systematisch an Tieren oder Menschen auf sein Missbrauchspotential untersucht. Während die klinischen Studien keine Tendenz zu Drogensuchtverhalten aufzeigten, waren diese Beobachtungen nicht systematisch und es ist nicht möglich, auf der Grundlage dieser begrenzten Erfahrung vorherzusagen, in welchem Ausmaß ein ZNS-aktives Medikament missbraucht, abgezweigt, und/oder nach der Vermarktung missbraucht werden. Folglich sollten Patienten sorgfältig auf Drogenmissbrauch in der Vorgeschichte untersucht werden, und solche Patienten sollten engmaschig auf Anzeichen von RISPERDAL®-Missbrauch oder -Missbrauch (z. B. Entwicklung einer Toleranz, Dosiserhöhungen, Suchtverhalten) beobachtet werden.
Abhängigkeit
RISPERDAL® wurde nicht systematisch an Tieren oder Menschen auf sein Potenzial für Toleranz oder körperliche Abhängigkeit untersucht.
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose
Ältere Patienten mit demenzbedingter Psychose, die mit Antipsychotika behandelt werden, haben ein erhöhtes Sterberisiko. Analysen von 17 placebokontrollierten Studien (modale Dauer von 10 Wochen), hauptsächlich bei Patienten, die atypische Antipsychotika einnahmen, ergaben ein Todesrisiko bei mit Arzneimitteln behandelten Patienten, das zwischen dem 1,6- und 1,7-fachen des Todesrisikos bei mit Placebo behandelten Patienten lag. Im Verlauf einer typischen 10-wöchigen kontrollierten Studie betrug die Todesrate bei mit Arzneimitteln behandelten Patienten etwa 4,5 %, verglichen mit einer Rate von etwa 2,6 % in der Placebogruppe. Obwohl die Todesursachen unterschiedlich waren, schienen die meisten Todesfälle entweder kardiovaskulärer (z. B. Herzinsuffizienz, plötzlicher Tod) oder infektiöser (z. B. Lungenentzündung) Natur zu sein. Beobachtungsstudien deuten darauf hin, dass die Behandlung mit konventionellen Antipsychotika, ähnlich wie bei atypischen Antipsychotika, die Sterblichkeit erhöhen kann. Inwieweit die Befunde einer erhöhten Sterblichkeit in Beobachtungsstudien auf das Antipsychotikum im Gegensatz zu einigen Merkmalen der Patienten zurückgeführt werden können, ist nicht klar.
In zwei von vier placebokontrollierten Studien mit älteren Patienten mit demenzbedingter Psychose wurde bei Patienten, die mit Furosemid plus RISPERDAL® behandelt wurden, eine höhere Sterblichkeitsrate beobachtet als bei Patienten, die mit RISPERDAL® allein oder mit Placebo plus Furosemid behandelt wurden. Es wurde kein pathologischer Mechanismus identifiziert, um diesen Befund zu erklären, und es wurde kein konsistentes Muster für die Todesursache beobachtet.
RISPERDAL® (Risperidon) ist nicht für die Behandlung von Demenz-assoziierter Psychose zugelassen [vgl Eingerahmte Warnung ].
Zerebrovaskuläre Nebenwirkungen, einschließlich Schlaganfall, bei älteren Patienten mit demenzbedingter Psychose
Zerebrovaskuläre Nebenwirkungen (z. B. Schlaganfall, transitorische ischämische Attacke), einschließlich Todesfälle, wurden bei Patienten (Durchschnittsalter 85 Jahre; Bereich 73–97) in Studien zu Risperidon bei älteren Patienten mit demenzbedingter Psychose berichtet. In placebokontrollierten Studien traten bei mit Risperidon behandelten Patienten im Vergleich zu mit Placebo behandelten Patienten signifikant häufiger zerebrovaskuläre Nebenwirkungen auf. RISPERDAL® ist nicht für die Behandlung von Patienten mit demenzbedingter Psychose zugelassen. [sehen Eingerahmte Warnung und Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose ]
Malignes neuroleptisches Syndrom
Antipsychotische Arzneimittel, einschließlich RISPERDAL®, können einen potenziell tödlichen Symptomkomplex hervorrufen, der als malignes neuroleptisches Syndrom (NMS) bezeichnet wird. Klinische Manifestationen von NMS umfassen Hyperpyrexie, Muskelrigidität, veränderten Geisteszustand und autonome Instabilität (unregelmäßiger Puls oder Blutdruck, Tachykardie, Schwitzen und Herzrhythmusstörungen). Zusätzliche Anzeichen können erhöhte Kreatinphosphokinase (CPK), Myoglobinurie, Rhabdomyolyse und akutes Nierenversagen sein.
Die diagnostische Beurteilung von Patienten mit diesem Syndrom ist kompliziert. Um zu einer Diagnose zu gelangen, ist es wichtig, Fälle zu identifizieren, in denen das klinische Erscheinungsbild sowohl eine schwere medizinische Erkrankung (z. B. Lungenentzündung, systemische Infektion usw.) als auch unbehandelte oder unzureichend behandelte extrapyramidale Zeichen und Symptome (EPS) umfasst. Andere wichtige Erwägungen bei der Differentialdiagnose umfassen die zentrale anticholinerge Toxizität, Hitzschlag, Drogenfieber und primäre Pathologie des Zentralnervensystems.
Das Management von NMS sollte umfassen: (1) sofortiges Absetzen von Antipsychotika und anderen Arzneimitteln, die für eine gleichzeitige Therapie nicht unbedingt erforderlich sind; (2) intensive symptomatische Behandlung und medizinische Überwachung; und (3) Behandlung aller begleitenden schwerwiegenden medizinischen Probleme, für die spezifische Behandlungen verfügbar sind. Es gibt keine allgemeine Übereinstimmung über spezifische pharmakologische Behandlungsschemata für unkompliziertes NMS.
Wenn ein Patient nach Genesung von NMS eine antipsychotische medikamentöse Behandlung benötigt, sollte die mögliche Wiederaufnahme der medikamentösen Therapie sorgfältig erwogen werden. Der Patient sollte sorgfältig überwacht werden, da Rezidive von NMS berichtet wurden.
Tardive Dyskinesie
Bei Patienten, die mit Antipsychotika behandelt werden, kann sich ein Syndrom potenziell irreversibler, unwillkürlicher, dyskinetischer Bewegungen entwickeln. Es wird angenommen, dass das Risiko der Entwicklung einer tardiven Dyskinesie und die Wahrscheinlichkeit, dass sie irreversibel wird, mit der Dauer der Behandlung und der kumulativen Gesamtdosis der dem Patienten verabreichten Antipsychotika zunimmt. Das Syndrom kann sich jedoch, wenn auch viel seltener, nach relativ kurzen Behandlungszeiten bei niedrigen Dosen entwickeln.
Es gibt keine bekannte Behandlung für etablierte Fälle von tardiver Dyskinesie, obwohl das Syndrom teilweise oder vollständig zurückgehen kann, wenn die antipsychotische Behandlung abgesetzt wird. Die antipsychotische Behandlung selbst kann jedoch die Anzeichen und Symptome des Syndroms unterdrücken (oder teilweise unterdrücken) und dadurch möglicherweise den zugrunde liegenden Prozess maskieren. Die Auswirkung der symptomatischen Unterdrückung auf den Langzeitverlauf des Syndroms ist unbekannt.
In Anbetracht dieser Überlegungen ist RISPERDAL® so zu verschreiben, dass das Auftreten von tardiven Dyskinesien am wahrscheinlichsten minimiert wird. Eine chronische antipsychotische Behandlung sollte im Allgemeinen Patienten vorbehalten bleiben, die an einer chronischen Krankheit leiden, die: (1) bekanntermaßen auf Antipsychotika anspricht und (2) für die alternative, ebenso wirksame, aber möglicherweise weniger schädliche Behandlungen nicht verfügbar oder angemessen sind. Bei Patienten, die eine chronische Behandlung benötigen, sollte die niedrigste Dosis und die kürzeste Behandlungsdauer angestrebt werden, die ein zufriedenstellendes klinisches Ansprechen bewirken. Die Notwendigkeit einer Fortsetzung der Behandlung sollte regelmäßig überprüft werden.
Wenn bei einem mit RISPERDAL® behandelten Patienten Anzeichen und Symptome einer tardiven Dyskinesie auftreten, ist ein Absetzen des Arzneimittels in Betracht zu ziehen. Einige Patienten benötigen jedoch möglicherweise trotz des Syndroms eine Behandlung mit RISPERDAL®.
Stoffwechselveränderungen
Atypische Antipsychotika wurden mit Stoffwechselveränderungen in Verbindung gebracht, die das kardiovaskuläre/zerebrovaskuläre Risiko erhöhen können. Diese metabolischen Veränderungen umfassen Hyperglykämie, Dyslipidämie und Körpergewichtszunahme. Obwohl gezeigt wurde, dass alle Medikamente dieser Klasse einige metabolische Veränderungen hervorrufen, hat jedes Medikament sein eigenes spezifisches Risikoprofil.
Hyperglykämie und Diabetes mellitus
Bei Patienten, die mit atypischen Antipsychotika einschließlich RISPERDAL® behandelt wurden, wurde über Hyperglykämie und Diabetes mellitus, in einigen Fällen extrem und verbunden mit Ketoazidose oder hyperosmolarem Koma oder Tod, berichtet. Die Beurteilung des Zusammenhangs zwischen der atypischen Anwendung von Antipsychotika und Glukoseanomalien wird durch die Möglichkeit eines erhöhten Hintergrundrisikos für Diabetes mellitus bei Patienten mit Schizophrenie und die zunehmende Inzidenz von Diabetes mellitus in der Allgemeinbevölkerung erschwert. Angesichts dieser Confounder ist die Beziehung zwischen der atypischen Anwendung von Antipsychotika und Hyperglykämie-bedingten unerwünschten Ereignissen nicht vollständig geklärt. Epidemiologische Studien weisen jedoch auf ein erhöhtes Risiko behandlungsbedingter Hyperglykämie-bedingter unerwünschter Ereignisse bei Patienten hin, die mit atypischen Antipsychotika behandelt werden. Präzise Risikoschätzungen für Hyperglykämie-bedingte unerwünschte Ereignisse bei Patienten, die mit atypischen Antipsychotika behandelt werden, sind nicht verfügbar.
Patienten mit einer gesicherten Diagnose von Diabetes mellitus, die mit atypischen Antipsychotika, einschließlich RISPERDAL®, begonnen werden, sollten regelmäßig auf eine Verschlechterung der Glukosekontrolle überwacht werden. Patienten mit Risikofaktoren für Diabetes mellitus (z. B. Fettleibigkeit, Diabetes in der Familienanamnese), die eine Behandlung mit atypischen Antipsychotika, einschließlich RISPERDAL®, beginnen, sollten sich zu Beginn der Behandlung und in regelmäßigen Abständen während der Behandlung einer Nüchtern-Blutzuckermessung unterziehen. Jeder Patient, der mit atypischen Antipsychotika, einschließlich RISPERDAL®, behandelt wird, sollte auf Symptome einer Hyperglykämie einschließlich Polydipsie, Polyurie, Polyphagie und Schwäche überwacht werden. Patienten, die während der Behandlung mit atypischen Antipsychotika, einschließlich RISPERDAL®, Symptome einer Hyperglykämie entwickeln, sollten sich einer Nüchtern-Blutzuckermessung unterziehen. In einigen Fällen klang die Hyperglykämie ab, wenn das atypische Antipsychotikum, einschließlich RISPERDAL®, abgesetzt wurde; Einige Patienten benötigten jedoch trotz Absetzens von RISPERDAL® eine Fortsetzung der antidiabetischen Behandlung.
Gepoolte Daten aus drei doppelblinden, placebokontrollierten Schizophrenie-Studien und vier doppelblinden, placebokontrollierten Studien mit bipolarer Monotherapie sind in Tabelle 2 dargestellt.
In längerfristigen, kontrollierten und unkontrollierten Studien war RISPERDAL® mit einer mittleren Veränderung des Glukosespiegels von +2,8 mg/dl in Woche 24 (n=151) und +4,1 mg/dl in Woche 48 (n=50) verbunden.
Daten aus der placebokontrollierten 3- bis 6-wöchigen Studie bei Kindern und Jugendlichen mit Schizophrenie (13-17 Jahre), bipolarer Manie (10-17 Jahre) oder autistischer Störung (5-17 Jahre) sind in Tabelle 3 dargestellt.
In längerfristigen, unkontrollierten, offenen pädiatrischen Verlängerungsstudien war RISPERDAL® mit einer mittleren Veränderung des Nüchternglukosespiegels von +5,2 mg/dl in Woche 24 (n=119) verbunden.
Dyslipidämie
Bei Patienten, die mit atypischen Antipsychotika behandelt wurden, wurden unerwünschte Veränderungen der Lipide beobachtet.
Gepoolte Daten aus 7 placebokontrollierten, 3- bis 8-wöchigen Studien mit fester oder flexibler Dosierung bei erwachsenen Probanden mit Schizophrenie oder bipolarer Manie sind in Tabelle 4 dargestellt.
In längerfristigen, kontrollierten und unkontrollierten Studien war RISPERDAL® mit einer mittleren Veränderung von (a) Nicht-Nüchtern-Cholesterin von +4,4 mg/dl in Woche 24 (n=231) und +5,5 mg/dl in Woche 48 ( n = 86); und (b) Nicht-Nüchtern-Triglyceride von +19,9 mg/dl in Woche 24 (n=52).
Gepoolte Daten aus 3 placebokontrollierten, 3- bis 6-wöchigen Studien mit fester Dosierung bei Kindern und Jugendlichen mit Schizophrenie (13-17 Jahre), bipolarer Manie (10-17 Jahre) oder autistischer Störung (5 -17 Jahre) sind in Tabelle 5 dargestellt.
In längerfristigen, unkontrollierten, offenen pädiatrischen Verlängerungsstudien war RISPERDAL® mit einer mittleren Veränderung von (a) Nüchterncholesterin von +2,1 mg/dl in Woche 24 (n=114); (b) Nüchtern-LDL von -0,2 mg/dl in Woche 24 (n=103); (c) Nüchtern-HDL von +0,4 mg/dL in Woche 24 (n=103); und (d) Nüchtern-Triglyceride von +6,8 mg/dL in Woche 24 (n=120).
Gewichtszunahme
Bei atypischer Anwendung von Antipsychotika wurde eine Gewichtszunahme beobachtet. Eine klinische Überwachung des Gewichts wird empfohlen.
Daten zu mittleren Veränderungen des Körpergewichts und zum Anteil der Probanden, die ein Gewichtszunahmekriterium von 7 % oder mehr des Körpergewichts erfüllen, aus 7 placebokontrollierten, 3- bis 8-wöchigen Studien mit fester oder flexibler Dosierung bei erwachsenen Probanden mit Schizophrenie oder bipolare Manie sind in Tabelle 6 dargestellt.
In längerfristigen, kontrollierten und unkontrollierten Studien war RISPERDAL® mit einer mittleren Gewichtsveränderung von +4,3 kg in Woche 24 (n=395) und +5,3 kg in Woche 48 (n=203) verbunden.
Daten zu mittleren Veränderungen des Körpergewichts und zum Anteil der Probanden, die das Kriterium einer Körpergewichtszunahme von ≥ 7 % erfüllen, aus neun placebokontrollierten, 3- bis 8-wöchigen Studien mit fester Dosierung bei Kindern und Jugendlichen mit Schizophrenie (13-17 Lebensjahr), bipolare Manie (10-17 Jahre), autistische Störung (5-17 Jahre) oder andere psychiatrische Störungen (5-17 Jahre) sind in Tabelle 7 aufgeführt.
In längerfristigen, unkontrollierten, offenen pädiatrischen Verlängerungsstudien war RISPERDAL® mit einer mittleren Gewichtsveränderung von +5,5 kg in Woche 24 (n=748) und +8,0 kg in Woche 48 (n=242) verbunden.
In einer offenen Langzeit-Verlängerungsstudie bei jugendlichen Patienten mit Schizophrenie wurde bei 14 % der Patienten eine Gewichtszunahme als behandlungsbedingte Nebenwirkung berichtet. Bei 103 jugendlichen Patienten mit Schizophrenie wurde nach 8-monatiger Behandlung mit RISPERDAL® eine mittlere Gewichtszunahme von 9,0 kg beobachtet. Der Großteil dieses Anstiegs wurde innerhalb der ersten 6 Monate beobachtet. Die durchschnittlichen Perzentile zu Studienbeginn bzw. nach 8 Monaten betrugen 56 und 72 für das Gewicht, 55 und 58 für die Körpergröße und 51 und 71 für den Body-Mass-Index.
In offenen Langzeitstudien (Studien an Patienten mit autistischer Störung oder anderen psychiatrischen Störungen) wurde eine mittlere Zunahme von 7,5 kg nach 12 Monaten RISPERDAL®-Behandlung beobachtet, was höher war als die erwartete normale Gewichtszunahme (ungefähr 3 auf 3,5 kg pro Jahr, angepasst an das Alter, basierend auf normativen Daten des Centers for Disease Control and Prevention). Der Großteil dieses Anstiegs trat innerhalb der ersten 6 Monate der Exposition gegenüber RISPERDAL® auf. Die durchschnittlichen Perzentile zu Studienbeginn bzw. nach 12 Monaten betrugen 49 und 60 für das Gewicht, 48 und 53 für die Körpergröße und 50 und 62 für den Body-Mass-Index.
In einer 3-wöchigen placebokontrollierten Studie bei Kindern und Jugendlichen mit akuten manischen oder gemischten Episoden einer Bipolar-I-Störung war die Zunahme des Körpergewichts in den RISPERDAL®-Gruppen höher als in der Placebo-Gruppe, jedoch nicht dosisabhängig (1,90 kg in RISPERDAL® 0,5-2,5 mg-Gruppe, 1,44 kg in der RISPERDAL® 3-6 mg-Gruppe und 0,65 kg in der Placebo-Gruppe). Ein ähnlicher Trend wurde bei der mittleren Veränderung des Body-Mass-Index gegenüber dem Ausgangswert beobachtet.
Bei der Behandlung von pädiatrischen Patienten mit RISPERDAL® für jegliche Indikation sollte die Gewichtszunahme im Vergleich zu der erwarteten bei normalem Wachstum beurteilt werden.
Hyperprolaktinämie
Wie bei anderen Arzneimitteln, die Dopamin-D2-Rezeptoren antagonisieren, erhöht RISPERDAL® die Prolaktinspiegel und die Erhöhung bleibt während der chronischen Verabreichung bestehen. RISPERDAL® wird mit höheren Prolaktinspiegeln in Verbindung gebracht als andere Antipsychotika.
Hyperprolaktinämie kann das hypothalamische GnRH unterdrücken, was zu einer verringerten Gonadotropin-Sekretion der Hypophyse führt. Dies wiederum kann die Fortpflanzungsfunktion hemmen, indem es die gonadale Steroidogenese sowohl bei weiblichen als auch bei männlichen Patienten beeinträchtigt. Galaktorrhoe, Amenorrhoe, Gynäkomastie und Impotenz wurden bei Patienten berichtet, die Prolaktin-erhöhende Präparate erhielten. Langanhaltende Hyperprolaktinämie in Verbindung mit Hypogonadismus kann sowohl bei weiblichen als auch bei männlichen Probanden zu einer verminderten Knochendichte führen.
Gewebekulturexperimente weisen darauf hin, dass ungefähr ein Drittel der menschlichen Brustkrebserkrankungen in vitro Prolaktin-abhängig sind, ein Faktor von potenzieller Bedeutung, wenn die Verschreibung dieser Arzneimittel bei einem Patienten mit zuvor festgestelltem Brustkrebs in Erwägung gezogen wird. In Karzinogenitätsstudien mit Risperidon, die an Mäusen und Ratten durchgeführt wurden, wurde eine Zunahme von Neoplasien der Hypophyse, der Brustdrüse und der Inselzellen der Bauchspeicheldrüse (Mamma-Adenokarzinome, Hypophysen- und Pankreas-Adenome) beobachtet [siehe Nichtklinische Toxikologie ]. Weder bisher durchgeführte klinische Studien noch epidemiologische Studien haben einen Zusammenhang zwischen der chronischen Verabreichung dieser Arzneimittelklasse und der Tumorentstehung beim Menschen gezeigt; Die verfügbaren Beweise gelten als zu begrenzt, um zum jetzigen Zeitpunkt schlüssig zu sein.
Orthostatische Hypotonie
RISPERDAL® kann eine orthostatische Hypotonie hervorrufen, die mit Schwindel, Tachykardie und bei manchen Patienten mit Synkopen einhergeht, insbesondere während der Anfangsphase der Dosistitration, was wahrscheinlich seine alpha-adrenergen antagonistischen Eigenschaften widerspiegelt. Synkopen wurden bei 0,2 % (6/2607) der mit RISPERDAL® behandelten Patienten in Studien der Phasen 2 und 3 bei Erwachsenen mit Schizophrenie berichtet. Das Risiko einer orthostatischen Hypotonie und Synkope kann minimiert werden, indem die Anfangsdosis bei normalen Erwachsenen auf insgesamt 2 mg (entweder einmal täglich oder 1 mg zweimal täglich) und bei älteren Patienten und Patienten mit eingeschränkter Nieren- oder Leberfunktion auf zweimal täglich 0,5 mg begrenzt wird [siehe DOSIERUNG UND ANWENDUNG ]. Bei Patienten, für die dies von Bedeutung ist, sollte eine Überwachung der orthostatischen Vitalfunktionen in Betracht gezogen werden. Bei Auftreten von Hypotonie sollte eine Dosisreduktion in Betracht gezogen werden. RISPERDAL® sollte bei Patienten mit bekannter Herz-Kreislauf-Erkrankung (Myokardinfarkt oder Ischämie in der Vorgeschichte, Herzinsuffizienz oder Reizleitungsstörungen), zerebrovaskulärer Erkrankung und Zuständen, die Patienten für Hypotonie prädisponieren würden, z. B. Dehydratation und Hypovolämie, mit besonderer Vorsicht angewendet werden. Bei gleichzeitiger Anwendung von RISPERDAL® und blutdrucksenkenden Medikamenten wurde eine klinisch signifikante Hypotonie beobachtet.
Leukopenie, Neutropenie und Agranulozytose
Klasseneffekt
In klinischen Studien und/oder Erfahrungen nach der Markteinführung wurden Fälle von Leukopenie/Neutropenie berichtet, die in zeitlichem Zusammenhang mit Antipsychotika, einschließlich RISPERDAL®, standen. Agranulozytose wurde ebenfalls berichtet.
Mögliche Risikofaktoren für Leukopenie/Neutropenie sind eine vorbestehende niedrige Anzahl weißer Blutkörperchen (WBC) und eine arzneimittelinduzierte Leukopenie/Neutropenie in der Vorgeschichte. Bei Patienten mit klinisch signifikant niedrigen WBC oder arzneimittelinduzierter Leukopenie/Neutropenie in der Anamnese sollte das komplette Blutbild (CBC) während der ersten Behandlungsmonate häufig überwacht werden, und ein Absetzen von RISPERDAL® sollte bei den ersten Anzeichen einer klinisch signifikanter Rückgang der WBC in Abwesenheit anderer ursächlicher Faktoren.
Patienten mit klinisch signifikanter Neutropenie sollten sorgfältig auf Fieber oder andere Symptome oder Anzeichen einer Infektion überwacht und unverzüglich behandelt werden, wenn solche Symptome oder Anzeichen auftreten. Patienten mit schwerer Neutropenie (absolute Neutrophilenzahl
Potenzial für kognitive und motorische Beeinträchtigungen
Schläfrigkeit war eine häufig berichtete Nebenwirkung im Zusammenhang mit der Behandlung mit RISPERDAL®, insbesondere wenn sie durch direkte Befragung der Patienten festgestellt wurde. Diese Nebenwirkung ist dosisabhängig, und in einer Studie, bei der eine Checkliste zum Nachweis von Nebenwirkungen verwendet wurde, berichteten 41 % der Hochdosis-Patienten (RISPERDAL® 16 mg/Tag) über Somnolenz im Vergleich zu 16 % der Placebo-Patienten.
Die direkte Befragung ist sensibler für die Erkennung von Nebenwirkungen als die Spontanmeldung, bei der 8 % der Patienten mit RISPERDAL® 16 mg/Tag und 1 % der Placebo-Patienten Somnolenz als Nebenwirkung angaben. Da RISPERDAL® das Urteilsvermögen, Denkvermögen oder motorische Fähigkeiten beeinträchtigen kann, sollten Patienten davor gewarnt werden, gefährliche Maschinen, einschließlich Autos, zu bedienen, bis sie hinreichend sicher sind, dass die RISPERDAL®-Therapie sie nicht beeinträchtigt.
Krampfanfälle
In Premarketing-Tests bei erwachsenen Patienten mit Schizophrenie traten bei 0,3 % (9/2607) der mit RISPERDAL® behandelten Patienten Krampfanfälle auf, zwei in Verbindung mit Hyponatriämie. RISPERDAL® sollte bei Patienten mit Krampfanfällen in der Vorgeschichte mit Vorsicht angewendet werden.
Dysphagie
Dysmotilität und Aspiration des Ösophagus wurden mit dem Gebrauch von Antipsychotika in Verbindung gebracht. Aspirationspneumonie ist eine häufige Ursache für Morbidität und Mortalität bei Patienten mit fortgeschrittener Alzheimer-Demenz. RISPERDAL® und andere Antipsychotika sollten bei Patienten mit einem Risiko für Aspirationspneumonie mit Vorsicht angewendet werden. [sehen Eingerahmte Warnung und Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose ]
Priapismus
Priapismus wurde während der Postmarketing-Überwachung berichtet. Ein schwerer Priapismus kann einen chirurgischen Eingriff erfordern.
Regulierung der Körpertemperatur
Eine Störung der Regulierung der Körpertemperatur wurde Antipsychotika zugeschrieben. Sowohl Hyperthermie als auch Hypothermie wurden im Zusammenhang mit der oralen Anwendung von RISPERDAL® berichtet. Bei der Verschreibung für Patienten, die extremen Temperaturen ausgesetzt sind, ist Vorsicht geboten.
Patienten mit Phenylketonurie
Informieren Sie die Patienten darüber, dass RISPERDAL® M-TAB® oral zerfallende Tabletten Phenylalanin enthalten. Phenylalanin ist ein Bestandteil von Aspartam. Jede 4 mg RISPERDAL® M-TAB® oral zerfallende Tablette enthält 0,84 mg Phenylalanin; jede 3 mg RISPERDAL® MTAB® oral zerfallende Tablette enthält 0,63 mg Phenylalanin; jede 2 mg RISPERDAL® M-TAB® oral zerfallende Tablette enthält 0,42 mg Phenylalanin; jede 1 mg RISPERDAL® M-TAB® Tablette zum Einnehmen enthält 0,28 mg Phenylalanin; und jede 0,5 mg RISPERDAL® M-TAB® oral auflösende Tablette enthält 0,14 mg Phenylalanin.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Karzinogenitätsstudien wurden an Swiss-Albino-Mäusen und Wistar-Ratten durchgeführt. Risperidon wurde Mäusen 18 Monate lang und Ratten 25 Monate lang in Dosen von 0,63 mg/kg, 2,5 mg/kg und 10 mg/kg über die Nahrung verabreicht. Diese Dosen entsprechen ungefähr dem 2-, 9- und 38-fachen der maximal empfohlenen Humandosis (MRHD) für Schizophrenie von 16 mg/Tag auf einer mg/kg-Basis oder dem 0,2-, 0,75- und 3-fachen der MRHD (Mäuse) oder 0,4. 1,5- und 6-mal die MRHD (Ratten) auf Basis von mg/m² Körperoberfläche. Bei männlichen Mäusen wurde keine maximal verträgliche Dosis erreicht. Es gab statistisch signifikante Zunahmen bei Adenomen der Hypophyse, Adenomen der endokrinen Bauchspeicheldrüse und Adenokarzinomen der Brustdrüse. Die folgende Tabelle fasst die Vielfachen der Humandosis auf mg/m² (mg/kg)-Basis zusammen, bei der diese Tumoren auftraten.
Es wurde gezeigt, dass Antipsychotika den Prolaktinspiegel bei Nagetieren chronisch erhöhen. Die Prolaktinspiegel im Serum wurden während der Karzinogenitätsstudien mit Risperidon nicht gemessen; Messungen während subchronischer Toxizitätsstudien zeigten jedoch, dass Risperidon bei Mäusen und Ratten die Serum-Prolaktinspiegel um das 5- bis 6-fache erhöhte, wenn dieselben Dosen in den Karzinogenitätsstudien verwendet wurden. Bei Nagern wurde nach chronischer Verabreichung anderer Antipsychotika eine Zunahme von Neoplasien der Brustdrüse, der Hypophyse und des endokrinen Pankreas festgestellt, die als Prolaktin-vermittelt angesehen wird. Die Relevanz der Befunde von Prolaktin-vermittelten endokrinen Tumoren bei Nagern für das Humanrisiko ist nicht bekannt [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Mutagenese
Im Ames-Genmutationstest, im Maus-Lymphom-Assay, im In-vitro-Ratten-Hepatozyten-DNA-Reparatur-Assay, im In-vivo-Mikrokerntest an Mäusen und im geschlechtsgebundenen rezessiven Letaltest bei Drosophila wurden keine Hinweise auf ein mutagenes oder klastogenes Potenzial von Risperidon gefunden , oder der Chromosomenaberrationstest in menschlichen Lymphozyten oder Eierstockzellen des chinesischen Hamsters.
Beeinträchtigung der Fruchtbarkeit
Risperidon (0,16 bis 5 mg/kg) hat in drei Reproduktionsstudien (zwei Segment-I- und eine Mehrgenerationenstudie) bei Wistar-Ratten bei Dosierungen, die dem 0,1- bis 3-Fachen der maximal empfohlenen Humandosis (MRHD) entsprachen, die Paarung, aber nicht die Fertilität beeinträchtigt auf Basis von mg/m² Körperoberfläche. Die Wirkung schien bei Weibchen aufzutreten, da in der Segment-I-Studie, in der nur Männchen behandelt wurden, kein beeinträchtigtes Paarungsverhalten festgestellt wurde. In einer subchronischen Studie an Beagle-Hunden, in denen Risperidon oral in Dosen von 0,31 bis 5 mg/kg verabreicht wurde, waren die Spermienmotilität und -konzentration bei Dosen, die das 0,6- bis 10-fache der MRHD auf Basis von mg/m² Körperoberfläche betrugen, verringert. Bei denselben Dosen wurden auch dosisabhängige Abnahmen des Serumtestosterons festgestellt. Serumtestosteron und Spermienparameter erholten sich teilweise, blieben aber nach Absetzen der Behandlung erniedrigt. Eine No-Effect-Dosierung konnte weder bei Ratten noch bei Hunden festgestellt werden.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschaftskategorie C
Zusammenfassung der Risiken
Es wurden keine angemessenen und gut kontrollierten Studien mit RISPERDAL 1 mg bei schwangeren Frauen durchgeführt. Neugeborene, die während des dritten Trimenons der Schwangerschaft Antipsychotika (einschließlich RISPERDAL®) ausgesetzt waren, haben ein erhöhtes Risiko für extrapyramidale und/oder Entzugssymptome nach der Entbindung. In embryo-fötalen Studien an Ratten und Kaninchen gab es beim 0,4- bis 6-Fachen der MHRD keinen Anstieg der Inzidenz von Missbildungen. In peripostnatalen Studien an Ratten wurde bei allen Dosen eine erhöhte Sterblichkeit der Jungtiere festgestellt. RISPERDAL® sollte während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen das potenzielle Risiko für den Fötus rechtfertigt.
Klinische Überlegungen
Fetale/neonatale Nebenwirkungen
Überwachen Sie Neugeborene, die extrapyramidale oder Entzugserscheinungen zeigen. Einige Neugeborene erholen sich ohne spezielle Behandlung innerhalb von Stunden oder Tagen; andere erfordern möglicherweise einen längeren Krankenhausaufenthalt.
Daten
Menschliche Daten
Es liegen Berichte über Agitiertheit, Hypertonie, Hypotonie, Tremor, Somnolenz, Atemnot und Fütterstörungen bei Neugeborenen vor, die im dritten Trimenon in utero Antipsychotika ausgesetzt waren. Diese Komplikationen haben unterschiedliche Schweregrade; Während in einigen Fällen die Symptome selbstlimitierend waren, mussten die Neugeborenen in anderen Fällen auf der Intensivstation betreut und länger im Krankenhaus behandelt werden.
Es gab einen Fall von Agenesie des Corpus callosum bei einem Säugling, der in utero Risperidon ausgesetzt war. Der kausale Zusammenhang mit der RISPERDAL®-Therapie ist nicht bekannt.
Tierdaten
Das teratogene Potenzial von Risperidon wurde in drei Segment-II-Studien an Sprague-Dawley- und Wistar-Ratten (0,63-10 mg/kg oder das 0,4- bis 6-fache der maximal empfohlenen Humandosis [MRHD] auf Basis von mg/m² Körperoberfläche) und untersucht in einer Segment-II-Studie an Neuseeland-Kaninchen (0,31-5 mg/kg oder 0,4- bis 6-fache MRHD auf Basis von mg/m² Körperoberfläche). Es gab keine teratogenen Wirkungen bei den Nachkommen von Ratten oder Kaninchen, denen das 0,4- bis 6-fache der MRHD, bezogen auf mg/m² Körperoberfläche, verabreicht wurde. In drei Reproduktionsstudien an Ratten (zwei Segment-III- und eine Mehrgenerationenstudie) kam es bei Dosierungen von 0,16-5 mg/kg oder dem 0,1- bis 3-fachen der MRHD bei einer mg/l m² Körperoberfläche Basis. Es ist nicht bekannt, ob diese Todesfälle auf eine direkte Auswirkung auf die Föten oder Jungtiere oder auf Auswirkungen auf die Muttertiere zurückzuführen waren.
Es gab keine No-Effect-Dosis für eine erhöhte Sterblichkeit von Rattenjungen. In einer Segment-III-Studie kam es bei einer Dosis von 2,5 mg/kg oder dem 1,5-fachen der MRHD auf Basis von mg/m² Körperoberfläche zu einer Zunahme totgeborener Rattenwelpen. In einer Cross-Fostering-Studie an Wistar-Ratten wurden toxische Wirkungen auf den Fötus oder die Jungtiere beobachtet, was durch eine Abnahme der Anzahl lebender Jungtiere und eine Zunahme der Anzahl toter Jungtiere bei der Geburt (Tag 0) und eine Abnahme belegt wurde im Geburtsgewicht bei Welpen von mit Arzneimitteln behandelten Muttertieren. Darüber hinaus gab es bis Tag 1 eine Zunahme der Todesfälle bei den Welpen von mit Arzneimitteln behandelten Muttertieren, unabhängig davon, ob die Welpen von einer Fremdfamilie stammten oder nicht. Risperidon schien auch das mütterliche Verhalten zu beeinträchtigen, da die Körpergewichtszunahme und das Überleben der Jungtiere (von Tag 1 bis 4 der Laktation) bei Jungtieren, die zur Kontrolle geboren, aber von mit Arzneimitteln behandelten Muttertieren aufgezogen wurden, verringert waren. Diese Wirkungen wurden alle bei einer getesteten Dosis von Risperidon festgestellt, dh 5 mg/kg oder das 3-fache der MRHD auf Basis von mg/m² Körperoberfläche.
Risperidon wird bei Rattenwelpen über die Plazenta übertragen.
Arbeit und Lieferung
Die Wirkung von RISPERDAL® auf Wehen und Entbindung beim Menschen ist nicht bekannt.
Stillende Mutter
Risperidon und 9-Hydroxyrisperidon sind in der Muttermilch vorhanden. Aufgrund der Möglichkeit schwerwiegender Nebenwirkungen von Risperidon bei gestillten Säuglingen sollte eine Entscheidung getroffen werden, ob das Stillen oder das Arzneimittel abgesetzt werden soll, wobei die Bedeutung des Arzneimittels für die Mutter zu berücksichtigen ist.
Pädiatrische Verwendung
Zugelassene pädiatrische Indikationen
Schizophrenie
Die Wirksamkeit und Sicherheit von RISPERDAL® bei der Behandlung von Schizophrenie wurde bei 417 Jugendlichen im Alter von 13 – 17 Jahren in zwei doppelblinden, kontrollierten Kurzzeitstudien (6 bzw. 8 Wochen) nachgewiesen [siehe INDIKATIONEN UND VERWENDUNG , NEBENWIRKUNGEN , und Klinische Studien ]. Zusätzliche Informationen zur Sicherheit und Wirksamkeit wurden auch in einer langfristigen (6-monatigen) unverblindeten Verlängerungsstudie bei 284 dieser jugendlichen Patienten mit Schizophrenie bewertet.
Sicherheit und Wirksamkeit von RISPERDAL® bei Kindern unter 13 Jahren mit Schizophrenie wurden nicht nachgewiesen.
Bipolare I-Störung
Die Wirksamkeit und Sicherheit von RISPERDAL® bei der Kurzzeitbehandlung von akuten manischen oder gemischten Episoden im Zusammenhang mit einer Bipolar-I-Störung bei 169 Kindern und Jugendlichen im Alter von 10 bis 17 Jahren wurde in einem doppelblinden, placebokontrollierten 3 -wöchige Testversion [siehe INDIKATIONEN UND VERWENDUNG , NEBENWIRKUNGEN , und Klinische Studien ].
Sicherheit und Wirksamkeit von RISPERDAL® bei Kindern unter 10 Jahren mit bipolarer Störung wurden nicht nachgewiesen.
Autistische Störung
Die Wirksamkeit und Sicherheit von RISPERDAL® bei der Behandlung von Reizbarkeit im Zusammenhang mit autistischen Störungen wurden in zwei 8-wöchigen, doppelblinden, placebokontrollierten Studien mit 156 Kindern und Jugendlichen im Alter von 5 bis 16 Jahren nachgewiesen [siehe INDIKATIONEN UND VERWENDUNG , NEBENWIRKUNGEN und Klinische Studien ]. Zusätzliche Sicherheitsinformationen wurden auch in einer Langzeitstudie bei Patienten mit autistischer Störung oder in Kurz- und Langzeitstudien bei mehr als 1200 pädiatrischen Patienten mit anderen psychiatrischen Störungen als autistischer Störung, Schizophrenie oder bipolarer Manie, die ähnlich waren, bewertet Alter und Gewicht, und die ähnliche Dosierungen von RISPERDAL® erhielten wie Patienten, die wegen Reizbarkeit im Zusammenhang mit einer autistischen Störung behandelt wurden.
Eine dritte Studie war eine 6-wöchige, multizentrische, randomisierte, doppelblinde, placebokontrollierte Studie mit fester Dosis zur Bewertung der Wirksamkeit und Sicherheit einer niedrigeren als der empfohlenen Dosis von Risperidon bei Personen im Alter von 5 bis 17 Jahren mit autistischer Störung und damit verbundene Reizbarkeit und damit verbundene Verhaltenssymptome. Es gab zwei gewichtsbasierte Fixdosen von Risperidon (hochdosiert und niedrigdosiert). Die hohe Dosis betrug 1,25 mg pro Tag für Patienten mit einem Gewicht von 20 bis 45 kg. Die niedrige Dosis betrug 0,125 mg pro Tag für Patienten mit einem Gewicht von 20 bis 45 kg. Die Studie zeigte die Wirksamkeit von hochdosiertem Risperidon, jedoch keine Wirksamkeit von niedrigdosiertem Risperidon.
Nebenwirkungen bei pädiatrischen Patienten
Tardive Dyskinesie
In klinischen Studien mit 1885 mit RISPERDAL® behandelten Kindern und Jugendlichen wurde bei 2 (0,1 %) Patienten eine tardive Dyskinesie berichtet, die nach Absetzen der RISPERDAL®-Behandlung verschwand [siehe auch WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Gewichtszunahme
Während der Behandlung mit RISPERDAL® wurde bei Kindern und Jugendlichen eine Gewichtszunahme beobachtet. Während der Behandlung wird eine klinische Überwachung des Gewichts empfohlen.
Die Daten stammen aus placebokontrollierten Kurzzeitstudien und unkontrollierten Langzeitstudien bei pädiatrischen Patienten (im Alter von 5 bis 17 Jahren) mit Schizophrenie, bipolarer Störung, autistischer Störung oder anderen psychiatrischen Störungen. In den Kurzzeitstudien (3 bis 8 Wochen) betrug die mittlere Gewichtszunahme bei den mit RISPERDAL® behandelten Patienten 2 kg, verglichen mit 0,6 kg bei den mit Placebo behandelten Patienten. In diesen Studien hatten etwa 33 % der RISPERDAL®-Gruppe eine Gewichtszunahme von > 7 %, verglichen mit 7 % in der Placebogruppe. In längerfristigen, unkontrollierten, offenen pädiatrischen Studien betrug die mittlere Gewichtszunahme 5,5 kg in Woche 24 und 8 kg in Woche 48 [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Schläfrigkeit
Somnolenz wurde häufig in placebokontrollierten klinischen Studien an pädiatrischen Patienten mit autistischer Störung beobachtet. Die meisten Fälle waren leicht oder mittelschwer. Diese Ereignisse traten meistens früh auf, wobei die höchste Inzidenz während der ersten beiden Behandlungswochen auftrat, und waren vorübergehend mit einer medianen Dauer von 16 Tagen. Somnolenz war die am häufigsten beobachtete Nebenwirkung in der klinischen Studie zu bipolarer Störung bei Kindern und Jugendlichen sowie in den Schizophrenie-Studien bei Jugendlichen. Wie in den Studien zu autistischen Störungen festgestellt wurde, traten diese Nebenwirkungen meistens früh auf und waren von vorübergehender Dauer [siehe NEBENWIRKUNGEN ]. Patienten mit anhaltender Somnolenz können von einer Änderung des Dosierungsschemas profitieren [siehe DOSIERUNG UND ANWENDUNG ].
Hyperprolaktinämie
RISPERDAL® erhöht nachweislich den Prolaktinspiegel bei Kindern und Jugendlichen sowie bei Erwachsenen [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]. In doppelblinden, placebokontrollierten Studien von bis zu 8 Wochen Dauer bei Kindern und Jugendlichen (im Alter von 5 bis 17 Jahren) mit autistischer Störung oder anderen psychiatrischen Störungen als autistischer Störung, Schizophrenie oder bipolarer Manie erhielten 49 % der Patienten RISPERDAL ® hatte im Vergleich zu 2 % der Patienten, die Placebo erhielten, erhöhte Prolaktinspiegel. In ähnlicher Weise hatten in placebokontrollierten Studien mit Kindern und Jugendlichen (im Alter von 10 bis 17 Jahren) mit bipolarer Störung oder Jugendlichen (im Alter von 13 bis 17 Jahren) mit Schizophrenie 82–87 % der Patienten, die RISPERDAL® erhielten, im Vergleich dazu erhöhte Prolaktinspiegel bei 3-7 % der Placebo-Patienten. Die Anstiege waren dosisabhängig und in allen Indikationen bei Frauen im Allgemeinen größer als bei Männern.
In klinischen Studien mit 1885 Kindern und Jugendlichen wurde Galaktorrhö bei 0,8 % der mit RISPERDAL® behandelten Patienten und Gynäkomastie bei 2,3 % der mit RISPERDAL® behandelten Patienten berichtet.
Wachstum und sexuelle Reifung
Die Langzeitwirkung von RISPERDAL® auf das Wachstum und die Geschlechtsreife bei Kindern und Jugendlichen wurde noch nicht vollständig untersucht.
Jugendtierstudien
Jugendliche Hunde wurden 40 Wochen lang mit oralen Risperidon-Dosen von 0,31, 1,25 oder 5 mg/kg/Tag behandelt. Bei einer Dosis ohne Wirkung von 0,31 mg/kg/Tag wurde eine verringerte Knochenlänge und -dichte beobachtet. Diese Dosis führte zu Plasmaspiegeln (AUC) von Risperidon plus seinem aktiven Metaboliten Paliperidon (9-Hydroxy-Risperidon), die denen bei Kindern und Jugendlichen ähnelten, die die maximal empfohlene Dosis für den Menschen (MRHD) von 6 mg/Tag erhielten. Darüber hinaus wurde bei allen Dosen sowohl bei Männern als auch bei Frauen eine Verzögerung der Geschlechtsreife beobachtet. Die oben genannten Wirkungen zeigten bei Frauen nach einer 12-wöchigen arzneimittelfreien Erholungsphase wenig oder keine Reversibilität.
In einer Studie, in der juvenile Ratten im Alter von 12 bis 50 Tagen mit oralem Risperidon behandelt wurden, wurde eine reversible Beeinträchtigung der Leistungsfähigkeit in einem Lern- und Gedächtnistest nur bei weiblichen Tieren mit einer Dosis ohne Wirkung von 0,63 mg/kg beobachtet /Tag. Diese Dosis führte zu Plasmaspiegeln (AUC) von Risperidon plus Paliperidon, die etwa halb so hoch waren wie die beim Menschen bei der MRHD beobachteten. Bis zur höchsten testbaren Dosis (1,25 mg/kg/Tag) wurden keine anderen konsistenten Wirkungen auf das neurologische Verhalten oder die Fortpflanzungsentwicklung beobachtet. Diese Dosis führte zu Plasmaspiegeln (AUC) von Risperidon plus Paliperidon, die etwa zwei Drittel der beim Menschen bei der MRHD beobachteten entsprachen.
Geriatrische Verwendung
Klinische Studien mit RISPERDAL® zur Behandlung von Schizophrenie schlossen keine ausreichende Anzahl von Patienten ab 65 Jahren ein, um festzustellen, ob sie anders ansprechen als jüngere Patienten. Andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt. Im Allgemeinen wird für einen älteren Patienten eine niedrigere Anfangsdosis empfohlen, was eine verminderte pharmakokinetische Clearance bei älteren Patienten sowie eine größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und einer Begleiterkrankung oder einer anderen medikamentösen Therapie widerspiegelt [siehe KLINISCHE PHARMAKOLOGIE und DOSIERUNG UND ANWENDUNG ]. Während ältere Patienten eine größere Neigung zu orthostatischer Hypotonie aufweisen, kann das Risiko bei älteren Patienten minimiert werden, indem die Anfangsdosis auf 0,5 mg zweimal täglich begrenzt wird, gefolgt von einer vorsichtigen Titration [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Bei Patienten, für die dies von Bedeutung ist, sollte eine Überwachung der orthostatischen Vitalfunktionen in Betracht gezogen werden.
Dieses Medikament wird im Wesentlichen über die Nieren ausgeschieden, und das Risiko toxischer Reaktionen auf dieses Medikament kann bei Patienten mit eingeschränkter Nierenfunktion größer sein. Da ältere Patienten eher eine eingeschränkte Nierenfunktion haben, sollte die Dosis sorgfältig ausgewählt werden und es kann sinnvoll sein, die Nierenfunktion zu überwachen [siehe DOSIERUNG UND ANWENDUNG ].
Nierenfunktionsstörung
Bei Patienten mit mittelschwerer bis schwerer (Clcr 59 bis 15 ml/min) Nierenerkrankung verringerte sich die Clearance der Summe von Risperidon und seinem aktiven Metaboliten im Vergleich zu jungen gesunden Probanden um 60 %. RISPERDAL®-Dosen sollten bei Patienten mit Nierenerkrankungen reduziert werden [siehe DOSIERUNG UND ANWENDUNG ].
Leberfunktionsstörung
Während die Pharmakokinetik von Risperidon bei Probanden mit Lebererkrankungen mit denen bei jungen gesunden Probanden vergleichbar war, war die mittlere freie Fraktion von Risperidon im Plasma aufgrund der verringerten Konzentration von sowohl Albumin als auch α1-Säure-Glykoprotein um etwa 35 % erhöht. RISPERDAL®-Dosen sollten bei Patienten mit Lebererkrankungen reduziert werden [siehe DOSIERUNG UND ANWENDUNG ].
Patienten mit Parkinson-Krankheit oder Lewy-Körper-Demenz
Patienten mit Parkinson-Krankheit oder Demenz mit Lewy-Körperchen können eine erhöhte Empfindlichkeit gegenüber RISPERDAL® erfahren. Manifestationen können Verwirrung, Benommenheit, posturale Instabilität mit häufigen Stürzen, extrapyramidale Symptome und klinische Merkmale umfassen, die mit einem malignen neuroleptischen Syndrom übereinstimmen.
ÜBERDOSIS
Menschliche Erfahrung
Die Erfahrungen vor der Markteinführung umfassten acht Berichte über eine akute Überdosierung von RISPERDAL® mit geschätzten Dosen zwischen 20 und 300 mg ohne Todesfälle. Im Allgemeinen handelte es sich bei den gemeldeten Anzeichen und Symptomen um solche, die auf eine Verstärkung der bekannten pharmakologischen Wirkungen des Arzneimittels zurückzuführen waren, dh Schläfrigkeit und Sedierung, Tachykardie und Hypotonie sowie extrapyramidale Symptome. Ein Fall mit einer geschätzten Überdosierung von 240 mg war mit Hyponatriämie, Hypokaliämie, verlängertem QT und erweitertem QRS verbunden. Ein weiterer Fall mit einer geschätzten Überdosis von 36 mg war mit einem Krampfanfall verbunden.
Erfahrungen nach der Markteinführung umfassen Berichte über eine akute Überdosierung von RISPERDAL® mit geschätzten Dosen von bis zu 360 mg. Im Allgemeinen sind die am häufigsten berichteten Anzeichen und Symptome solche, die aus einer Übertreibung der bekannten pharmakologischen Wirkungen des Arzneimittels resultieren, dh Schläfrigkeit, Sedierung, Tachykardie, Hypotonie und extrapyramidale Symptome. Andere Nebenwirkungen, die seit der Markteinführung im Zusammenhang mit einer Überdosierung von RISPERDAL® berichtet wurden, umfassen verlängertes QT-Intervall und Krämpfe. Torsade de Pointes wurde im Zusammenhang mit einer kombinierten Überdosierung von RISPERDAL® und Paroxetin berichtet.
Management einer Überdosierung
Wenden Sie sich für die aktuellsten Informationen zum Umgang mit einer Überdosierung von RISPERDAL® an eine zertifizierte Giftnotrufzentrale (1-800-222-1222 oder www.poison.org). Sorgen Sie für unterstützende Pflege, einschließlich einer engmaschigen medizinischen Überwachung und Überwachung. Die Behandlung sollte aus allgemeinen Maßnahmen bestehen, die bei der Behandlung einer Überdosierung mit jedem Arzneimittel angewendet werden. Berücksichtigen Sie die Möglichkeit einer mehrfachen Überdosierung des Arzneimittels. Sorgen Sie für ausreichende Atemwege, Sauerstoffversorgung und Belüftung. Überwachen Sie den Herzrhythmus und die Vitalfunktionen. Verwenden Sie unterstützende und symptomatische Maßnahmen. Es gibt kein spezifisches Gegenmittel für RISPERDAL®.
KONTRAINDIKATIONEN
RISPERDAL® ist kontraindiziert bei Patienten mit bekannter Überempfindlichkeit gegenüber Risperidon oder Paliperidon oder einem der sonstigen Bestandteile der RISPERDAL®-Formulierung. Überempfindlichkeitsreaktionen, einschließlich anaphylaktischer Reaktionen und Angioödem, wurden bei Patienten berichtet, die mit Risperidon und bei Patienten behandelt wurden, die mit Paliperidon behandelt wurden. Paliperidon ist ein Metabolit von Risperidon.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Der Wirkungsmechanismus von RISPERDAL® bei Schizophrenie ist unbekannt. Es wurde jedoch vorgeschlagen, dass die therapeutische Aktivität des Medikaments bei Schizophrenie durch eine Kombination aus Dopamin-Typ-2- (D2) und Serotonin-Typ-2- (5HT2)-Rezeptorantagonismus vermittelt werden könnte. Die klinische Wirkung von RISPERDAL® ergibt sich aus den kombinierten Konzentrationen von Risperidon und seinem Hauptmetaboliten 9-Hydroxyrisperidon [vgl Wirkmechanismus ]. Antagonismus an anderen Rezeptoren als D2 und 5HT2 [vgl Pharmakokinetik ] kann einige der anderen Wirkungen von RISPERDAL® erklären.
Pharmakodynamik
RISPERDAL® ist ein selektiver monoaminerger Antagonist mit hoher Affinität (Ki von 0,12 bis 7,3 nM) für Serotonin Typ 2 (5HT2), Dopamin Typ 2 (D2), α1- und α2-adrenerge und H1-histaminerge Rezeptoren. RISPERDAL® wirkt als Antagonist an anderen Rezeptoren, jedoch mit geringerer Potenz. RISPERDAL® hat eine geringe bis mäßige Affinität (Ki von 47 bis 253 nM) für die Serotonin-Rezeptoren 5HT1C, 5HT1D und 5HT1A, eine schwache Affinität (Ki von 620 bis 800 nM) für die Dopamin-D1- und Haloperidol-sensitive Sigma-Stelle und keine Affinität (wenn bei Konzentrationen > 10-5 M getestet) auf cholinerge Muscarin- oder β1- und β2-adrenerge Rezeptoren.
Pharmakokinetik
Absorption
Risperidon wird gut resorbiert. Die absolute orale Bioverfügbarkeit von Risperidon beträgt 70 % (VK = 25 %). Die relative orale Bioverfügbarkeit von Risperidon aus einer Tablette beträgt 94 % (CV = 10 %) im Vergleich zu einer Lösung.
Pharmakokinetische Studien haben gezeigt, dass RISPERDAL® M-TAB® oral zerfallende Tabletten und RISPERDAL® Lösung zum Einnehmen bioäquivalent zu RISPERDAL® Tabletten sind.
Die Plasmakonzentrationen von Risperidon, seinem Hauptmetaboliten 9-Hydroxyrisperidon und Risperidon plus 9-Hydroxyrisperidon sind über den Dosierungsbereich von 1 bis 16 mg täglich (0,5 bis 8 mg zweimal täglich) dosisproportional. Nach oraler Gabe einer Lösung oder Tablette traten mittlere maximale Plasmakonzentrationen von Risperidon nach etwa 1 Stunde auf. Spitzenkonzentrationen von 9-Hydroxyrisperidon traten bei schnellen Metabolisierern nach etwa 3 Stunden und bei langsamen Metabolisierern nach 17 Stunden auf. Steady-State-Konzentrationen von Risperidon werden bei schnellen Metabolisierern innerhalb von 1 Tag erreicht und sollten bei langsamen Metabolisierern nach etwa 5 Tagen erreicht werden. Steady-State-Konzentrationen von 9-Hydroxyrisperidon werden in 5-6 Tagen erreicht (gemessen in schnellen Metabolisierern).
Food-Effekt
Nahrung beeinflusst weder die Rate noch das Ausmaß der Resorption von Risperidon. Daher kann RISPERDAL® mit oder ohne Mahlzeiten verabreicht werden.
Verteilung
Risperidon wird schnell verteilt. Das Verteilungsvolumen beträgt 1-2 l/kg. Im Plasma wird Risperidon an Albumin und α1-saures Glykoprotein gebunden. Die Plasmaproteinbindung von Risperidon beträgt 90 % und die seines Hauptmetaboliten 9-Hydroxyrisperidon 77 %. Weder Risperidon noch 9-Hydroxyrisperidon verdrängen sich gegenseitig von Plasmabindungsstellen. Hohe therapeutische Konzentrationen von Sulfamethazin (100 µg/ml), Warfarin (10 µg/ml) und Carbamazepin (10 µg/ml) verursachten nur einen leichten Anstieg der freien Fraktion von Risperidon bei 10 ng/ml und 9-Hydroxyrisperidon bei 50 ng /ml, Veränderungen von unbekannter klinischer Bedeutung.
Stoffwechsel
Risperidon wird weitgehend in der Leber metabolisiert. Der Hauptstoffwechselweg verläuft über die Hydroxylierung von Risperidon zu 9-Hydroxyrisperidon durch das Enzym CYP 2D6. Ein kleiner Stoffwechselweg ist die N-Dealkylierung. Der Hauptmetabolit, 9-Hydroxyrisperidon, hat eine ähnliche pharmakologische Aktivität wie Risperidon. Folglich ergibt sich die klinische Wirkung des Medikaments aus den kombinierten Konzentrationen von Risperidon plus 9-Hydroxyrisperidon.
CYP 2D6, auch Debrisoquin-Hydroxylase genannt, ist das Enzym, das für den Metabolismus vieler Neuroleptika, Antidepressiva, Antiarrhythmika und anderer Arzneimittel verantwortlich ist. CYP 2D6 unterliegt einem genetischen Polymorphismus (etwa 6 %–8 % der Kaukasier und ein sehr geringer Prozentsatz der Asiaten haben wenig oder keine Aktivität und sind „schwache Metabolisierer“) und einer Hemmung durch eine Vielzahl von Substraten und einigen Nicht-Substraten , insbesondere Chinidin. Extensive CYP 2D6-Metabolisierer wandeln Risperidon schnell in 9-Hydroxyrisperidon um, wohingegen schwache CYP 2D6-Metabolisierer es viel langsamer umwandeln. Obwohl schnelle Metabolisierer niedrigere Risperidon- und höhere 9-Hydroxyrisperidon-Konzentrationen aufweisen als langsame Metabolisierer, ist die Pharmakokinetik von Risperidon und 9-Hydroxyrisperidon kombiniert nach Einzel- und Mehrfachgabe bei schnellen und langsamen Metabolisierern ähnlich.
Risperidon kann zwei Arten von Arzneimittelwechselwirkungen unterliegen. Erstens stören Inhibitoren von CYP 2D6 die Umwandlung von Risperidon in 9-Hydroxyrisperidon [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ]. Dies tritt bei Chinidin auf, was im Wesentlichen allen Empfängern ein pharmakokinetisches Profil von Risperidon verleiht, das typisch für langsame Metabolisierer ist. Der therapeutische Nutzen und die Nebenwirkungen von Risperidon bei Patienten, die Chinidin erhielten, wurden nicht bewertet, aber Beobachtungen bei einer bescheidenen Anzahl (n≅70) von langsamen Metabolisierern, denen RISPERDAL® verabreicht wurde, deuten nicht auf wichtige Unterschiede zwischen langsamen und schnellen Metabolisierern hin. Zweitens kann die gleichzeitige Verabreichung bekannter Enzyminduktoren (z. B. Carbamazepin, Phenytoin, Rifampin und Phenobarbital) mit RISPERDAL® zu einer Abnahme der kombinierten Plasmakonzentrationen von Risperidon und 9-Hydroxyrisperidon führen [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ]. Es wäre auch möglich, dass Risperidon den Metabolismus anderer Arzneimittel, die durch CYP 2D6 metabolisiert werden, beeinflusst. Eine relativ schwache Bindung von Risperidon an das Enzym legt nahe, dass dies unwahrscheinlich ist [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
In-vitro-Studien weisen darauf hin, dass Risperidon ein relativ schwacher Inhibitor von CYP 2D6 ist. Daher ist nicht zu erwarten, dass RISPERDAL® die Clearance von Arzneimitteln, die über diesen enzymatischen Weg metabolisiert werden, wesentlich hemmt. In Arzneimittelwechselwirkungsstudien beeinflusste RISPERDAL® die Pharmakokinetik von Donepezil und Galantamin, die durch CYP 2D6 metabolisiert werden, nicht signifikant.
In-vitro-Studien zeigten, dass Arzneimittel, die von anderen CYP-Isoenzymen, einschließlich 1A1, 1A2, 2C9, 2C19 und 3A4, metabolisiert werden, nur schwache Inhibitoren des Risperidon-Metabolismus sind.
Ausscheidung
Risperidon und seine Metaboliten werden über den Urin und in viel geringerem Maße über die Faeces ausgeschieden. Wie durch eine Massenbilanzstudie einer oralen Einzeldosis von 1 mg 14C-Risperidon, die drei gesunden männlichen Freiwilligen als Lösung verabreicht wurde, gezeigt wurde, betrug die Gesamtwiederfindung der Radioaktivität nach 1 Woche 84 %, einschließlich 70 % im Urin und 14 % im Stuhl .
Die scheinbare Halbwertszeit von Risperidon betrug 3 Stunden (CV = 30 %) bei schnellen Metabolisierern und 20 Stunden (CV = 40 %) bei langsamen Metabolisierern. Die scheinbare Halbwertszeit von 9-Hydroxyrisperidon betrug etwa 21 Stunden (CV = 20 %) bei schnellen Metabolisierern und 30 Stunden (CV = 25 %) bei langsamen Metabolisierern. Die Pharmakokinetik von Risperidon und 9-Hydroxyrisperidon zusammen war nach Einzel- und Mehrfachdosen bei schnellen und langsamen Metabolisierern ähnlich, mit einer mittleren Eliminationshalbwertszeit von insgesamt etwa 20 Stunden.
Arzneimittelwechselwirkungsstudien
[Sehen WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Spezifische Populationen
Nieren- und Leberfunktionsstörung
[Sehen Verwendung in bestimmten Populationen ].
Alten
Bei gesunden älteren Probanden war die renale Clearance sowohl von Risperidon als auch von 9-Hydroxyrisperidon im Vergleich zu jungen gesunden Probanden verringert und die Eliminationshalbwertszeit verlängert. Die Dosierung sollte bei älteren Patienten entsprechend angepasst werden [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Pädiatrie
Die Pharmakokinetik von Risperidon und 9-Hydroxyrisperidon bei Kindern war nach Korrektur des Unterschieds im Körpergewicht ähnlich wie bei Erwachsenen.
Rassen- und Geschlechtseffekte
Es wurde keine spezifische pharmakokinetische Studie zur Untersuchung von Rassen- und Geschlechtseffekten durchgeführt, aber eine populationspharmakokinetische Analyse ergab keine wesentlichen Unterschiede in der Disposition von Risperidon aufgrund des Geschlechts (ob körpergewichtskorrigiert oder nicht) oder der Rasse.
Tiertoxikologie
Jugendliche Hunde wurden 40 Wochen lang mit oralen Risperidon-Dosen von 0,31, 1,25 oder 5 mg/kg/Tag behandelt. Bei einer Dosis ohne Wirkung von 0,31 mg/kg/Tag wurde eine verringerte Knochenlänge und -dichte beobachtet. Diese Dosis führte zu Plasma-AUC-Spiegeln von Risperidon plus seinem aktiven Metaboliten Paliperidon (9-Hydroxy-Risperidon), die denen bei Kindern und Jugendlichen ähnelten, die die maximal empfohlene Dosis für den Menschen (MRHD) von 6 mg/Tag erhielten. Darüber hinaus wurde bei allen Dosen sowohl bei Männern als auch bei Frauen eine Verzögerung der Geschlechtsreife beobachtet. Die oben genannten Wirkungen zeigten bei Frauen nach einer 12-wöchigen arzneimittelfreien Erholungsphase wenig oder keine Reversibilität.
In einer Studie, in der juvenile Ratten im Alter von 12 bis 50 Tagen mit oralem Risperidon behandelt wurden, wurde bei Weibchen nur bei einer Dosis ohne Wirkung von 0,63 mg/kg/Tag eine reversible Beeinträchtigung der Leistungsfähigkeit in einem Lern- und Gedächtnistest beobachtet . Diese Dosis führte zu Plasma-AUC-Spiegeln von Risperidon plus Paliperidon, die etwa halb so hoch waren wie die beim Menschen bei der MRHD beobachteten. Bis zur höchsten testbaren Dosis von 1,25 mg/kg/Tag wurden keine anderen konsistenten Wirkungen auf das neurologische Verhalten oder die Fortpflanzungsentwicklung beobachtet. Diese Dosis führte zu Plasma-AUC-Spiegeln von Risperidon plus Paliperidon, die etwa zwei Drittel der beim Menschen bei der MRHD beobachteten entsprachen.
Klinische Studien
Schizophrenie
Erwachsene
Kurzfristige Wirksamkeit
Die Wirksamkeit von RISPERDAL® bei der Behandlung von Schizophrenie wurde in vier kontrollierten Kurzzeitstudien (4 bis 8 Wochen) mit stationären psychotischen Patienten nachgewiesen, die die DSM-III-R-Kriterien für Schizophrenie erfüllten.
In diesen Studien wurden mehrere Instrumente zur Bewertung psychiatrischer Anzeichen und Symptome verwendet, darunter die Brief Psychiatric Rating Scale (BPRS), eine aus mehreren Elementen bestehende Bestandsaufnahme der allgemeinen Psychopathologie, die traditionell zur Bewertung der Auswirkungen einer medikamentösen Behandlung bei Schizophrenie verwendet wird. Das BPRS-Psychose-Cluster (konzeptionelle Desorganisation, halluzinatorisches Verhalten, Misstrauen und ungewöhnliche Gedankeninhalte) gilt als besonders nützliche Untergruppe zur Beurteilung aktiv psychotischer schizophrener Patienten. Eine zweite traditionelle Bewertung, der Clinical Global Impression (CGI), spiegelt den Eindruck eines erfahrenen Beobachters, der mit den Manifestationen der Schizophrenie vollständig vertraut ist, über den klinischen Gesamtzustand des Patienten wider. Zusätzlich wurden die Positive and Negative Syndrome Scale (PANSS) und die Scale for Assessing Negative Symptoms (SANS) verwendet.
Die Ergebnisse der Versuche folgen:
Langzeitwirksamkeit
In einer längerfristigen Studie wurden 365 erwachsene ambulante Patienten, die überwiegend die DSM-IV-Kriterien für Schizophrenie erfüllten und die seit mindestens 4 Wochen unter einer antipsychotischen Medikation klinisch stabil waren, zu RISPERDAL® (2-8 mg/Tag) oder zu einem Wirkstoff randomisiert Komparator für 1 bis 2 Jahre Beobachtung auf Rückfall. Patienten, die RISPERDAL® erhielten, erlebten in diesem Zeitraum eine signifikant längere Zeit bis zum Rückfall im Vergleich zu denen, die das aktive Vergleichspräparat erhielten.
Pädiatrie
Die Wirksamkeit von RISPERDAL® bei der Behandlung von Schizophrenie bei Jugendlichen im Alter von 13–17 Jahren wurde in zwei doppelblinden, kontrollierten Kurzzeitstudien (6 und 8 Wochen) nachgewiesen. Alle Patienten erfüllten die DSM-IV-Diagnosekriterien für Schizophrenie und hatten zum Zeitpunkt der Aufnahme eine akute Episode. In der ersten Studie (Studie Nr. 1) wurden die Patienten in eine von drei Behandlungsgruppen randomisiert: RISPERDAL® 1–3 mg/Tag (n = 55, mittlere modale Dosis = 2,6 mg), RISPERDAL® 4–6 mg/Tag ( n = 51, mittlere modale Dosis = 5,3 mg) oder Placebo (n = 54). In der zweiten Studie (Studie Nr. 2) wurden die Patienten randomisiert entweder RISPERDAL® 0,15–0,6 mg/Tag (n = 132, mittlere modale Dosis = 0,5 mg) oder RISPERDAL® 1,5–6 mg/Tag (n = 125, Mittelwert) zugeteilt modale Dosis = 4 mg). In allen Fällen wurde die Studienmedikation mit 0,5 mg/Tag begonnen (mit Ausnahme der 0,15–0,6 mg/Tag-Gruppe in Studie Nr. 2, wo die Anfangsdosis 0,05 mg/Tag betrug) und um ungefähr auf den Zieldosisbereich titriert Tag 7. Anschließend wurde die Dosis bis Tag 14 auf die maximal tolerierte Dosis innerhalb des Zieldosisbereichs erhöht. Die primäre Wirksamkeitsvariable in allen Studien war die mittlere Veränderung des PANSS-Gesamtscores gegenüber dem Ausgangswert.
Die Ergebnisse der Studien zeigten die Wirksamkeit von RISPERDAL® in allen Dosisgruppen von 1–6 mg/Tag im Vergleich zu Placebo, gemessen an der signifikanten Verringerung des PANSS-Gesamtscores. Die Wirksamkeit des primären Parameters in der Gruppe mit 1–3 mg/Tag war vergleichbar mit der Gruppe mit 4–6 mg/Tag in Studie Nr. 1 und ähnlich der Wirksamkeit, die in der Gruppe mit 1,5–6 mg/Tag in Studie Nr. 2 gezeigt wurde . In Studie Nr. 2 war die Wirksamkeit in der Gruppe mit 1,5–6 mg/Tag statistisch signifikant höher als in der Gruppe mit 0,15–0,6 mg/Tag. Dosen über 3 mg/Tag zeigten keinen Trend zu größerer Wirksamkeit.
Bipolare Manie - Monotherapie
Erwachsene
Die Wirksamkeit von RISPERDAL® bei der Behandlung von akuten manischen oder gemischten Episoden wurde in zwei placebokontrollierten Kurzzeitstudien (3 Wochen) bei Patienten nachgewiesen, die die DSM-IV-Kriterien für Bipolar-I-Störung mit manischen oder gemischten Episoden erfüllten. Diese Studien schlossen Patienten mit oder ohne psychotische Merkmale ein.
Das primäre Bewertungsinstrument, das in diesen Studien zur Bewertung manischer Symptome verwendet wurde, war die Young Mania Rating Scale (YMRS), eine 11-Punkte-Skala, die von Klinikern bewertet wird und traditionell verwendet wird, um den Grad der manischen Symptomatik (Reizbarkeit, störendes/aggressives Verhalten, Schlaf, erhöhter Blutdruck) zu bewerten Stimmung, Sprache, erhöhte Aktivität, sexuelles Interesse, Sprach-/Denkstörung, Gedankeninhalt, Aussehen und Einsicht) in einem Bereich von 0 (keine manischen Züge) bis 60 (maximale Punktzahl). Der primäre Endpunkt in diesen Studien war die Veränderung des YMRS-Gesamtscores gegenüber dem Ausgangswert. Die Ergebnisse der Versuche folgen:
Pädiatrie
Die Wirksamkeit von RISPERDAL® bei der Behandlung von Manie bei Kindern oder Jugendlichen mit Bipolar-I-Störung wurde in einer 3-wöchigen, randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studie mit Patienten im Alter von 10 bis 17 Jahren nachgewiesen Erleben einer manischen oder gemischten Episode einer Bipolar-I-Störung. Die Patienten wurden in eine von drei Behandlungsgruppen randomisiert: RISPERDAL® 0,5–2,5 mg/Tag (n = 50, mittlere modale Dosis = 1,9 mg), RISPERDAL® 3–6 mg/Tag (n = 61, mittlere modale Dosis = 4,7 mg). ) oder Placebo (n = 58). In allen Fällen wurde die Studienmedikation mit 0,5 mg/Tag begonnen und bis Tag 7 auf den Zieldosisbereich titriert, mit weiteren Dosiserhöhungen bis zur maximal verträglichen Dosis innerhalb des Zieldosisbereichs bis Tag 10. Das primäre Bewertungsinstrument, das zur Bewertung verwendet wurde Die Wirksamkeit war in dieser Studie die mittlere Veränderung des YMRS-Gesamtscores gegenüber dem Ausgangswert.
Die Ergebnisse dieser Studie zeigten die Wirksamkeit von RISPERDAL® in beiden Dosisgruppen im Vergleich zu Placebo, gemessen an der signifikanten Verringerung des YMRS-Gesamtwerts. Die Wirksamkeit in Bezug auf den primären Parameter in der Dosisgruppe mit 3–6 mg/Tag war vergleichbar mit der Dosisgruppe mit 0,5–2,5 mg/Tag. Dosen über 2,5 mg/Tag zeigten keinen Trend zu größerer Wirksamkeit.
Bipolare Manie – Zusatztherapie mit Lithium oder Valproat
Die Wirksamkeit von RISPERDAL® mit gleichzeitiger Gabe von Lithium oder Valproat bei der Behandlung von akuten manischen oder gemischten Episoden wurde in einer kontrollierten Studie mit erwachsenen Patienten nachgewiesen, die die DSM-IV-Kriterien für eine Bipolar-I-Störung erfüllten. Diese Studie umfasste Patienten mit oder ohne psychotische Merkmale und mit oder ohne Rapid-Cycling-Verlauf.
Reizbarkeit im Zusammenhang mit autistischer Störung
Kurzfristige Wirksamkeit
Die Wirksamkeit von RISPERDAL® bei der Behandlung von Reizbarkeit im Zusammenhang mit autistischen Störungen wurde in zwei 8-wöchigen, placebokontrollierten Studien mit Kindern und Jugendlichen (im Alter von 5 bis 16 Jahren), die die DSM-IV-Kriterien für autistische Störungen erfüllten, nachgewiesen. Über 90 % dieser Probanden waren unter 12 Jahre alt und die meisten wogen über 20 kg (16-104,3 kg).
Die Wirksamkeit wurde anhand von zwei Bewertungsskalen bewertet: der Aberrant Behavior Checklist (ABC) und der Clinical Global Impression – Change (CGI-C)-Skala. Der primäre Endpunkt in beiden Studien war die Veränderung von der Baseline zum Endpunkt in der Subskala Reizbarkeit des ABC (ABC-I). Die ABC-I-Subskala misst die emotionalen und Verhaltenssymptome von Autismus, einschließlich Aggression gegenüber anderen, vorsätzlicher Selbstverletzung, Wutausbrüchen und schnell wechselnden Stimmungen. Die CGI-C-Bewertung am Endpunkt war in einer der Studien ein co-primärer Endpunkt.
Die Ergebnisse dieser Versuche sind wie folgt:
Eine dritte Studie war eine 6-wöchige, multizentrische, randomisierte, doppelblinde, placebokontrollierte Studie mit fester Dosis zur Bewertung der Wirksamkeit und Sicherheit einer niedrigeren als der empfohlenen Dosis von Risperidon bei Probanden (N = 96) im Alter von 5 bis 17 Jahren volljährig mit autistischer Störung (definiert durch DSM-IV-Kriterien) und damit verbundener Reizbarkeit und damit verbundenen Verhaltenssymptomen. Ungefähr 77 % der Patienten waren jünger als 12 Jahre (Durchschnittsalter = 9) und 88 % waren männlich. Die meisten Patienten (73 %) wogen weniger als 45 kg (Durchschnittsgewicht = 40 kg). Ungefähr 90 % der Patienten waren vor Eintritt in die Studie Antipsychotika-naiv.
Es gab zwei gewichtsbasierte Fixdosen von Risperidon (hochdosiert und niedrigdosiert). Die hohe Dosis betrug 1,25 mg pro Tag für Patienten mit einem Gewicht von 20 bis 45 kg. Die niedrige Dosis betrug 0,125 mg pro Tag für Patienten mit einem Gewicht von 20 bis 45 kg. Die Dosis wurde einmal täglich morgens oder abends verabreicht, wenn eine Sedierung auftrat.
Der primäre Wirksamkeitsendpunkt war die durchschnittliche Veränderung des ABC-I-Scores der Aberrant Behavior Checklist – Irritability Subscale vom Ausgangswert bis zum Ende von Woche 6. Die Studie zeigte die Wirksamkeit von hochdosiertem Risperidon, gemessen an der durchschnittlichen ABC-Änderung -Ich Punkte. Es zeigte keine Wirksamkeit für niedrig dosiertes Risperidon. Die mittleren ABC-I-Scores zu Studienbeginn betrugen 29 in der Placebo-Gruppe (n = 35), 27 in der Risperidon-Niedrigdosis-Gruppe (n = 30) und 28 in der Risperidon-Hochdosis-Gruppe (n = 31). Die mittleren Veränderungen der ABC-I-Scores betrugen -3,5, -7,4 und -12,4 in der Placebo-, Niedrigdosis- bzw. Hochdosisgruppe. Die Ergebnisse in der Hochdosisgruppe waren statistisch signifikant (p
Langzeitwirksamkeit
Nach Abschluss der ersten 8-wöchigen Doppelblindstudie nahmen 63 Patienten an einer offenen Verlängerungsstudie teil, in der sie 4 oder 6 Monate lang mit RISPERDAL® behandelt wurden (je nachdem, ob sie in der Doppelblindstudie RISPERDAL® oder Placebo erhielten ). Während dieses offenen Behandlungszeitraums wurden die Patienten auf einer mittleren modalen Dosis von RISPERDAL® von 1,8–2,1 mg/Tag (entsprechend 0,05–0,07 mg/kg/Tag) gehalten.
Patienten, die ihr positives Ansprechen auf RISPERDAL® während der 4- bis 6-monatigen offenen Label-Behandlungsphase von durchschnittlich etwa 140 Tagen wurden randomisiert, um RISPERDAL® oder Placebo während einer 8-wöchigen, doppelblinden Absetzstudie zu erhalten (n=39 der 63 Patienten). Eine vorab geplante Zwischenanalyse der Daten von Patienten, die die Entzugsstudie abgeschlossen haben (n=32), die von einem unabhängigen Data Safety Monitoring Board durchgeführt wurde, zeigte eine signifikant niedrigere Rückfallrate in der RISPERDAL®-Gruppe im Vergleich zur Placebogruppe. Basierend auf den Zwischenanalyseergebnissen wurde die Studie aufgrund des Nachweises einer statistisch signifikanten Wirkung auf die Rückfallprävention beendet. Rückfall war definiert als ≥ 25 % Verschlechterung bei der letzten Bewertung der ABC-I-Subskala (in Bezug auf den Ausgangswert der randomisierten Absetzphase).
INFORMATIONEN ZUM PATIENTEN
Ärzten wird geraten, die folgenden Punkte mit Patienten, denen sie RISPERDAL® verschreiben, und ihren Betreuern zu besprechen:
Orthostatische Hypotonie
Informieren Sie Patienten und Pflegekräfte über das Risiko einer orthostatischen Hypotonie, insbesondere während der anfänglichen Dosistitration [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Beeinträchtigung der kognitiven und motorischen Leistung
Informieren Sie Patienten und Pflegekräfte darüber, dass RISPERDAL® das Urteilsvermögen, Denkvermögen oder motorische Fähigkeiten beeinträchtigen kann. Vorsicht beim Bedienen gefährlicher Maschinen, einschließlich Autos, bis die Patienten hinreichend sicher sind, dass die RISPERDAL®-Therapie sie nicht nachteilig beeinflusst [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Schwangerschaft
Raten Sie Patientinnen und Pflegekräften, ihren Arzt zu benachrichtigen, wenn die Patientin während der Therapie schwanger wird oder eine Schwangerschaft beabsichtigt [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Pflege
Informieren Sie Patienten und Pflegekräfte darüber, dass Risperidon und sein aktiver Metabolit in der Muttermilch vorhanden sind; Es besteht die Möglichkeit schwerwiegender Nebenwirkungen von RISPERDAL® bei gestillten Säuglingen. Weisen Sie die Patienten darauf hin, dass die Entscheidung, ob sie das Stillen oder das Absetzen von RISPERDAL® beenden, die Bedeutung des Arzneimittels für den Patienten berücksichtigen sollte [siehe Verwendung in bestimmten Populationen ].
Begleitmedikation
Raten Sie Patienten und Pflegekräften, ihre Ärzte zu informieren, wenn der Patient verschreibungspflichtige oder rezeptfreie Medikamente einnimmt oder einzunehmen beabsichtigt, da die Möglichkeit von Wechselwirkungen besteht [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Alkohol
Weisen Sie die Patienten darauf hin, Alkohol während der Einnahme von RISPERDAL® zu vermeiden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Phenylketonurika
Informieren Sie Patienten mit Phenylketonurie und ihre Pflegekräfte darüber, dass RISPERDAL® M-TAB® im Mund auflösende Tabletten Phenylalanin enthalten. Phenylalanin ist ein Bestandteil von Aspartam. Jede 4 mg RISPERDAL® M-TAB® oral zerfallende Tablette enthält 0,84 mg Phenylalanin; jede 3 mg RISPERDAL® M-TAB® oral zerfallende Tablette enthält 0,63 mg Phenylalanin; jede 2 mg RISPERDAL® M-TAB® oral zerfallende Tablette enthält 0,42 mg Phenylalanin; jede 1 mg RISPERDAL® M-TAB® Tablette zum Einnehmen enthält 0,28 mg Phenylalanin; und jede 0,5 mg RISPERDAL® M-TAB® oral auflösende Tablette enthält 0,14 mg Phenylalanin [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Stoffwechselveränderungen
Informieren Sie Patienten und Betreuer darüber, dass die Behandlung mit RISPERDAL® mit Hyperglykämie und Diabetes mellitus, Dyslipidämie und Gewichtszunahme in Verbindung gebracht werden kann [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Tardive Dyskinesie
Informieren Sie Patienten und Pflegekräfte über das Risiko einer tardiven Dyskinesie [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Was ist Robaxin 500 mg und wie wird es angewendet?
Robaxin 500 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Muskelkrämpfen, die durch Schmerzen oder Verletzungen und Tetanus verursacht werden. Robaxin 500 mg kann allein oder zusammen mit anderen Medikamenten verwendet werden.
Welche Nebenwirkungen kann Robaxin 500 mg haben?
Robaxin 500 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Robaxin gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Robaxin. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
robaxin®/robaxin®- 750 (Methocarbamol-Tabletten, USP), ein Carbamat-Derivat von Guaifenesin, wirkt dämpfend auf das Zentralnervensystem (ZNS) mit sedierenden und muskuloskelettrelaxierenden Eigenschaften.
Der chemische Name von Methocarbamol lautet 3-(2-Methoxyphenoxy)-1,2-propandiol-1-carbamat und hat die Summenformel C11H15NO5. Sein Molekulargewicht beträgt 241,24. Die Strukturformel ist unten gezeigt.
Methocarbamol ist ein weißes Pulver, schwer löslich in Wasser und Chloroform, löslich in Alkohol (nur unter Erhitzen) und Propylenglykol und unlöslich in Benzol und n-Hexan.
robaxin® ist als hellorange, runde Filmtablette mit 500 mg Methocarbamol, USP, zur oralen Verabreichung erhältlich. Die enthaltenen inaktiven Inhaltsstoffe sind Maisstärke, FD&C Yellow 6, Hydroxypropylcellulose, Hypromellose, Magnesiumstearat, Polysorbat 20, Povidon, Propylenglykol, Saccharin-Natrium, Natriumlaurylsulfat, Natriumstärkeglykolat, Stearinsäure, Titandioxid.
robaxin®- 750 ist als orangefarbene, kapselförmige Filmtablette mit 750 mg Methocarbamol, USP, zur oralen Verabreichung erhältlich. Zusätzlich zu den in robaxin® enthaltenen Hilfsstoffen enthält robaxin® 750 auch D&C Yellow 10.
INDIKATIONEN
Die injizierbare Form von Methocarbamol ist als Ergänzung zu Ruhe, Physiotherapie und anderen Maßnahmen zur Linderung von Beschwerden in Verbindung mit akuten, schmerzhaften Erkrankungen des Bewegungsapparates indiziert. Die Wirkungsweise dieses Medikaments wurde nicht eindeutig identifiziert, kann aber mit seinen beruhigenden Eigenschaften zusammenhängen. Methocarbamol entspannt verspannte Skelettmuskeln beim Menschen nicht direkt.
DOSIERUNG UND ANWENDUNG
Nur zur intravenösen und intramuskulären Anwendung. Die Gesamtdosis für Erwachsene sollte 30 ml (3 Durchstechflaschen) pro Tag an mehr als 3 aufeinanderfolgenden Tagen nicht überschreiten, außer bei der Behandlung von Tetanus. Wenn der Zustand anhält, kann ein ähnlicher Kurs nach einem medikamentenfreien Intervall von 48 Stunden wiederholt werden. Dosierung und Häufigkeit der Injektion sollten auf der Schwere der zu behandelnden Erkrankung und dem festgestellten therapeutischen Ansprechen basieren.
Zur Linderung mittelgradiger Symptome kann eine Dosis von 1 Gramm (eine 10-ml-Durchstechflasche) ausreichend sein. Normalerweise muss diese Injektion nicht wiederholt werden, da die Verabreichung der oralen Form normalerweise die durch die Injektion eingeleitete Linderung aufrechterhält. In den schwersten Fällen oder bei postoperativen Zuständen, bei denen eine orale Verabreichung nicht möglich ist, können zusätzliche Dosen von 1 Gramm alle 8 Stunden bis zu einer Höchstdosis von 3 g/Tag an nicht mehr als 3 aufeinanderfolgenden Tagen wiederholt werden.
Anweisungen zur intravenösen Anwendung
ROBAXIN Injectable kann unverdünnt mit einer maximalen Geschwindigkeit von 3 ml pro Minute direkt in die Vene verabreicht werden. Es kann auch einem intravenösen Tropf einer Natriumchlorid-Injektion (sterile isotonische Natriumchloridlösung zur parenteralen Anwendung) oder einer fünfprozentigen Dextrose-Injektion (sterile 5-prozentige Dextrose-Lösung) hinzugefügt werden; Eine als Einzeldosis verabreichte Durchstechflasche sollte für die intravenöse Infusion nicht auf mehr als 250 ml verdünnt werden. NACH DEM VERMISCHEN MIT INFUSIONSFLÜSSIGKEITEN NICHT IM KÜHLBEREICH AUFBEWAHREN. Es ist darauf zu achten, dass eine vaskuläre Extravasation dieser hypertonen Lösung vermieden wird, die zu einer Thrombophlebitis führen kann. Es ist vorzuziehen, dass der Patient während und für mindestens 10 bis 15 Minuten nach der Injektion in einer liegenden Position ist.
Anweisungen zur intramuskulären Anwendung
Wenn die intramuskuläre Verabreichung angezeigt ist, sollten nicht mehr als fünf ml (eine halbe Durchstechflasche) in jede Gesäßregion injiziert werden. Die Injektionen können bei Bedarf in 8-Stunden-Intervallen wiederholt werden. Wenn eine zufriedenstellende Linderung der Symptome erreicht ist, kann sie normalerweise mit Tabletten aufrechterhalten werden.
Nicht für die subkutane Verabreichung empfohlen.
Besondere Gebrauchsanweisung bei Tetanus
Es gibt klinische Hinweise darauf, dass Methocarbamol eine positive Wirkung bei der Kontrolle der neuromuskulären Manifestationen von Tetanus haben kann. Es ersetzt jedoch nicht das übliche Verfahren mit Débridement, Tetanus-Antitoxin, Penicillin, Tracheotomie, Beachtung des Flüssigkeitshaushalts und unterstützender Pflege. ROBAXIN Injectable sollte dem Regime so bald wie möglich hinzugefügt werden.
Für Erwachsene
Injizieren Sie eine oder zwei Durchstechflaschen direkt in den Schlauch der zuvor eingeführten Verweilkanüle. Der Infusionsflasche können weitere 10 ml oder 20 ml hinzugefügt werden, sodass insgesamt bis zu 30 ml (drei Durchstechflaschen) als Anfangsdosis verabreicht werden (siehe VORSICHTSMASSNAHMEN ). Dieses Verfahren sollte alle sechs Stunden wiederholt werden, bis die Bedingungen das Einführen einer Magensonde zulassen. Zerkleinerte Methocarbamol-Tabletten, suspendiert in Wasser oder Kochsalzlösung, können dann durch diese Sonde verabreicht werden. Je nach Ansprechen des Patienten können orale Gesamtdosen von bis zu 24 Gramm erforderlich sein.
Für pädiatrische Patienten
Es wird eine Anfangsdosis von mindestens 15 mg/kg oder 500 mg/m² empfohlen. Diese Dosierung kann bei Bedarf alle sechs Stunden wiederholt werden. Die Gesamtdosis sollte 1,8 g/m² an 3 aufeinanderfolgenden Tagen nicht überschreiten. Die Erhaltungsdosis kann durch Injektion in einen Schlauch oder durch intravenöse Infusion mit einer geeigneten Menge Flüssigkeit verabreicht werden. Siehe Gebrauchsanweisung für die intravenöse Anwendung.
WIE GELIEFERT
ROBAXIN 500 mg Injektionslösung (100 mg/ml) geliefert in - 10-ml-Einzeldosis-Durchstechflaschen in Packungen mit 25 ( NDC 0641-6103-25).
Lagerung bei 20°- 25°C (68°- 77°F), Auslagerungen bis 15°- 30°C (59°- 86°F) zulässig.
Nicht aus Naturkautschuklatex hergestellt.
Um VERMUTLICHE NEBENWIRKUNGEN zu melden, kontaktieren Sie West-Ward Pharmaceuticals Corp. unter 1-877-845-0689 oder die FDA unter 1-800-FDA-1088 oder www.fda.gov/medwatch.
Rufen Sie für Produktanfragen 1-877-845-0689 an.
Hergestellt von: WEST-WARD A HIKMA COMPANY, Eatontown, NJ 07724 USA. Überarbeitet: Okt. 2017
NEBENWIRKUNGEN
Die folgenden Nebenwirkungen wurden gleichzeitig mit der Verabreichung von Methocarbamol berichtet. Einige Ereignisse können auf eine zu schnelle intravenöse Injektionsrate zurückzuführen sein.
Körper als Ganzes: Anaphylaktische Reaktion, angioneurotisches Ödem, Fieber, Kopfschmerzen
Herz-Kreislauf-System: Bradykardie, Flush, Hypotonie, Synkope, Thrombophlebitis
In den meisten Fällen von Synkopen kam es zu einer spontanen Genesung. In anderen Fällen wurden Epinephrin, injizierbare Steroide und/oder injizierbare Antihistaminika eingesetzt, um die Genesung zu beschleunigen.
Verdauungstrakt: Dyspepsie, Gelbsucht (einschließlich cholestatischer Gelbsucht), Übelkeit und Erbrechen
Hämisches und lymphatisches System: Leukopenie
Immunsystem: Überempfindlichkeitsreaktionen
Nervöses System: Amnesie, Verwirrtheit, Diplopie, Schwindel oder Benommenheit, Benommenheit, Schlaflosigkeit, leichte muskuläre Koordinationsstörungen, Nystagmus, Sedierung, Krampfanfälle (einschließlich Grand Mal), Schwindel
Das Auftreten von Krampfanfällen während der intravenösen Gabe von Methocarbamol wurde bei Patienten mit Anfallsleiden berichtet. Das psychische Trauma des Eingriffs könnte ein Faktor gewesen sein, der dazu beigetragen hat. Obwohl mehrere Beobachter von Erfolgen bei der Beendigung epileptiformer Anfälle mit ROBAXIN Injectable berichtet haben, wird seine Anwendung bei Patienten mit Epilepsie nicht empfohlen (siehe VORSICHTSMASSNAHMEN , Allgemein ).
Haut und besondere Sinne: Verschwommenes Sehen, Konjunktivitis, verstopfte Nase, metallischer Geschmack, Pruritus, Hautausschlag, Nesselsucht
Sonstiges: Schmerzen und Verschorfung an der Injektionsstelle
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Sehen WARNUNGEN UND VORSICHTSMASSNAHMEN für Wechselwirkungen mit ZNS-Medikamenten und Alkohol.
Methocarbamol kann die Wirkung von Pyridostigminbromid hemmen. Daher sollte Methocarbamol bei Patienten mit Myasthenia gravis, die Anticholinesterasemittel erhalten, mit Vorsicht angewendet werden.
Arzneimittel-/Labortest-Wechselwirkungen
Methocarbamol kann bei bestimmten Suchtests für 5-Hydroxyindolessigsäure (5-HIAA) unter Verwendung von Nitrosonaphthol-Reagenz und bei Suchtests für Urin-Vanillylmandelsäure (VMA) unter Verwendung der Gitlow-Methode eine Farbstörung verursachen.
WARNUNGEN
Da Methocarbamol eine allgemein dämpfende Wirkung auf das ZNS haben kann, sollten Patienten, die ROBAXIN Injectable erhalten, vor kombinierten Wirkungen mit Alkohol und anderen ZNS-dämpfenden Mitteln gewarnt werden.
Die sichere Anwendung von ROBAXIN Injectable im Hinblick auf mögliche Nebenwirkungen auf die fetale Entwicklung ist nicht erwiesen. Es gab sehr seltene Berichte über fötale und angeborene Anomalien nach In-utero-Exposition gegenüber Methocarbamol. Daher sollte ROBAXIN Injectable bei Frauen, die schwanger sind oder werden könnten, und insbesondere in der Frühschwangerschaft, nicht angewendet werden, es sei denn, der potenzielle Nutzen überwiegt nach Einschätzung des Arztes die möglichen Gefahren (siehe Abschnitt 4.4). VORSICHTSMASSNAHMEN , Schwangerschaft ).
Verwendung bei Aktivitäten, die geistige Wachsamkeit erfordern
Methocarbamol kann die geistigen und/oder körperlichen Fähigkeiten beeinträchtigen, die für die Ausführung gefährlicher Aufgaben wie das Bedienen von Maschinen oder das Führen eines Kraftfahrzeugs erforderlich sind. Patienten sollten vor dem Bedienen von Maschinen, einschließlich Kraftfahrzeugen, gewarnt werden, bis sie hinreichend sicher sind, dass die Methocarbamol-Therapie ihre Fähigkeit zur Ausübung solcher Tätigkeiten nicht beeinträchtigt.
VORSICHTSMASSNAHMEN
Allgemein
Wie bei anderen Wirkstoffen, die entweder intravenös oder intramuskulär verabreicht werden, sollten Dosis und Injektionsrate sorgfältig überwacht werden. Die Injektionsgeschwindigkeit sollte 3 ml pro Minute nicht überschreiten, dh eine 10-ml-Durchstechflasche in etwa drei Minuten. Da ROBAXIN 500 mg zur Injektion hypertonisch ist, muss eine Gefäßextravasation vermieden werden. Eine liegende Position verringert die Wahrscheinlichkeit von Nebenreaktionen.
In die Spritze aspiriertes Blut vermischt sich nicht mit der hypertonen Lösung. Dieses Phänomen tritt bei vielen anderen intravenösen Präparaten auf. Das Blut kann mit dem Methocarbamol injiziert werden, oder die Injektion kann gestoppt werden, wenn der Kolben das Blut erreicht, je nachdem, was der Arzt bevorzugt.
Die Gesamtdosis sollte 30 ml (drei Durchstechflaschen) pro Tag an mehr als drei aufeinanderfolgenden Tagen nicht überschreiten, außer bei der Behandlung von Tetanus.
Vorsicht ist geboten bei der Anwendung der injizierbaren Form bei Patienten mit vermuteten oder bekannten Anfallsleiden.
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Langzeitstudien zur Bewertung des kanzerogenen Potenzials von Methocarbamol wurden nicht durchgeführt. Es wurden keine Studien durchgeführt, um die Wirkung von Methocarbamol auf die Mutagenese oder sein Potenzial zur Beeinträchtigung der Fruchtbarkeit zu beurteilen.
Schwangerschaft
Teratogene Wirkungen
Schwangerschaftskategorie C
Reproduktionsstudien an Tieren wurden mit Methocarbamol nicht durchgeführt. Es ist auch nicht bekannt, ob Methocarbamol bei Verabreichung an eine schwangere Frau den Fötus schädigen oder die Fortpflanzungsfähigkeit beeinträchtigen kann. ROBAXIN 500 mg Injektionslösung sollte einer schwangeren Frau nur verabreicht werden, wenn dies eindeutig erforderlich ist.
Die sichere Anwendung von ROBAXIN Injectable im Hinblick auf mögliche Nebenwirkungen auf die fetale Entwicklung ist nicht erwiesen. Es liegen Berichte über fötale und angeborene Anomalien nach In-utero-Exposition gegenüber Methocarbamol vor. Daher sollte ROBAXIN 500 mg zur Injektion bei Frauen, die schwanger sind oder werden könnten, und insbesondere in der Frühschwangerschaft nicht angewendet werden, es sei denn, der mögliche Nutzen überwiegt nach Einschätzung des Arztes die möglichen Gefahren (siehe Abschnitt 4.4). WARNUNGEN ).
Stillende Mutter
Methocarbamol und/oder seine Metaboliten gehen in die Milch von Hunden über; es ist jedoch nicht bekannt, ob Methocarbamol oder seine Metaboliten in die Muttermilch übergehen. Da viele Arzneimittel in die Muttermilch ausgeschieden werden, ist Vorsicht geboten, wenn ROBAXIN 500 mg zur Injektion einer stillenden Frau verabreicht wird.
Pädiatrische Verwendung
Die Sicherheit und Wirksamkeit von ROBAXIN Injectable bei pädiatrischen Patienten wurde außer bei Tetanus nicht untersucht. Sehen DOSIERUNG UND ANWENDUNG , Besondere Anweisungen zum Anwendung bei Tetanus , Für pädiatrische Patienten .
ÜBERDOSIS
Zur akuten Toxizität von Methocarbamol liegen nur begrenzte Informationen vor. Eine Überdosierung von Methocarbamol erfolgt häufig in Verbindung mit Alkohol oder anderen ZNS-dämpfenden Mitteln und umfasst die folgenden Symptome: Übelkeit, Schläfrigkeit, verschwommenes Sehen, Hypotonie, Krampfanfälle und Koma. Nach der Markteinführung wurde über Todesfälle bei einer Überdosierung von Methocarbamol allein oder in Gegenwart anderer zentral dämpfender Mittel, Alkohol oder Psychopharmaka berichtet.
Behandlung
Die Behandlung einer Überdosierung umfasst eine symptomatische und unterstützende Behandlung. Zu den unterstützenden Maßnahmen gehören die Aufrechterhaltung ausreichender Atemwege, die Überwachung der Harnausscheidung und der Vitalfunktionen sowie die Verabreichung von intravenösen Flüssigkeiten, falls erforderlich. Der Nutzen der Hämodialyse bei der Behandlung einer Überdosierung ist nicht bekannt.
KONTRAINDIKATIONEN
ROBAXIN 500 mg zur Injektion sollte Patienten mit bekannter oder vermuteter Nierenerkrankung nicht verabreicht werden. Diese Vorsicht ist wegen des Vorhandenseins von Polyethylenglykol 300 im Fahrzeug erforderlich.
Es ist bekannt, dass eine viel größere Menge Polyethylenglycol 300 als in den empfohlenen Dosen von ROBAXIN Injectable vorhanden ist, bei Patienten mit eingeschränkter Nierenfunktion zu einer verstärkten vorbestehenden Azidose und Harnstoffretention führt. Obwohl die in diesem Präparat vorhandene Menge innerhalb der Sicherheitsgrenzen liegt, ist bei dieser Kontraindikation Vorsicht geboten.
ROBAXIN Injectable ist bei Patienten kontraindiziert, die gegen Methocarbamol oder einen der Injektionsbestandteile überempfindlich sind.
KLINISCHE PHARMAKOLOGIE
Der Wirkungsmechanismus von Methocarbamol beim Menschen ist nicht bekannt, kann aber auf eine allgemeine ZNS-Depression zurückzuführen sein. Es hat keine direkte Wirkung auf den kontraktilen Mechanismus der quergestreiften Muskulatur, der motorischen Endplatte oder der Nervenfaser.
Pharmakokinetik
Bei gesunden Probanden liegt die Plasmaclearance von Methocarbamol zwischen 0,20 und 0,80 l/h/kg, die mittlere Plasmaeliminationshalbwertszeit zwischen 1 und 2 Stunden und die Plasmaproteinbindung zwischen 46 % und 50 %.
Methocarbamol wird über Dealkylierung und Hydroxylierung metabolisiert. Auch eine Konjugation von Methocarbamol ist wahrscheinlich. Im Wesentlichen werden alle Metaboliten von Methocarbamol im Urin ausgeschieden. Kleine Mengen von unverändertem Methocarbamol werden auch mit dem Urin ausgeschieden.
Besondere Populationen
Alten
Die mittlere (± SD) Eliminationshalbwertszeit von Methocarbamol war bei älteren gesunden Probanden (mittleres (± SD) Alter 69 (± 4) Jahre) im Vergleich zu einem jüngeren (mittleres (± SD) Alter 53,3 (± 8,8 ) Jahre), gesunde Bevölkerung (1,5 (± 0,4) Stunden versus 1,1 (± 0,27) Stunden). Der Anteil an gebundenem Methocarbamol war bei älteren Probanden im Vergleich zu jüngeren Probanden leicht verringert (41 bis 43 % bzw. 46 bis 50 %).
Nierenbeeinträchtigt
Die Clearance von Methocarbamol war bei 8 nierengeschädigten Patienten unter Erhaltungs-Hämodialyse im Vergleich zu 17 gesunden Probanden um etwa 40 % reduziert, obwohl die mittlere (± SD) Eliminationshalbwertszeit in diesen beiden Gruppen ähnlich war (1,2 (± 0,6) versus 1,1 ( ± 0,3) Stunden).
Leberfunktionsstörung
Bei 8 Patienten mit Zirrhose infolge von Alkoholmissbrauch war die mittlere Gesamtclearance von Methocarbamol im Vergleich zu 8 normalen Probanden gleichen Alters und Gewichts um etwa 70 % reduziert. Die mittlere (±SD) Eliminationshalbwertszeit bei Patienten mit Zirrhose und gesunden Probanden betrug 3,38 (± 1,62) Stunden bzw. 1,11 (± 0,27) Stunden. Der Prozentsatz von an Plasmaproteine gebundenem Methocarbamol war auf etwa 40 bis 45 % verringert, verglichen mit 46 bis 50 % bei gesunden Probanden.
INFORMATIONEN ZUM PATIENTEN
Die Patienten sollten darauf hingewiesen werden, dass Methocarbamol Schläfrigkeit oder Schwindel verursachen kann, was ihre Fähigkeit zum Bedienen von Kraftfahrzeugen oder Maschinen beeinträchtigen kann.
Da Methocarbamol eine allgemeine ZNS-dämpfende Wirkung haben kann, sollten Patienten vor kombinierten Wirkungen mit Alkohol und anderen ZNS-dämpfenden Mitteln gewarnt werden.
Was ist Seroquel 100 mg und wie wird es angewendet?
Seroquel 50 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Schizophrenie, Bipolar-I-Störung, Manie; Bipolare Störung, depressive Episoden; Bipolare I-Störung, Wartung; und Major Depression. Seroquel 50 mg kann allein oder zusammen mit anderen Medikamenten verwendet werden.
Seroquel gehört zu einer Medikamentenklasse namens Antipsychotika der 2. Generation; Antimanische Wirkstoffe.
Es ist nicht bekannt, ob Seroquel 200 mg bei Kindern unter 12 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Seroquel 50 mg haben?
Seroquel kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Seroquel gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Seroquel. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
WARNUNG
ERHÖHTE STERBLICHKEIT BEI ÄLTEREN PATIENTEN MIT PSYCHOSE IM ZUSAMMENHANG MIT DEMENZ; und SELBSTMORDIGE GEDANKEN UND VERHALTENSWEISEN
Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose
Ältere Patienten mit demenzbedingter Psychose, die mit Antipsychotika behandelt werden, haben ein erhöhtes Sterberisiko [siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN]. SEROQUEL ist nicht für die Behandlung von Patienten mit demenzbedingter Psychose zugelassen [siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN].
Suizidgedanken und Verhaltensweisen
Antidepressiva erhöhten in Kurzzeitstudien das Risiko für Suizidgedanken und -verhalten bei Kindern, Jugendlichen und jungen Erwachsenen. Diese Studien zeigten keine Erhöhung des Risikos für Suizidgedanken und -verhalten bei Anwendung von Antidepressiva bei Patienten über 24 Jahren; bei Patienten im Alter von 65 Jahren und älter wurde das Risiko bei der Anwendung von Antidepressiva verringert [siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN].
Bei Patienten jeden Alters, die mit einer antidepressiven Therapie begonnen werden, ist eine genaue Überwachung auf eine Verschlechterung und das Auftreten von Suizidgedanken und -verhalten vorzunehmen. Informieren Sie Familien und Pflegekräfte über die Notwendigkeit einer genauen Beobachtung und Kommunikation mit dem verschreibenden Arzt [siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN].
SEROQUEL 50 mg ist nicht für die Anwendung bei pädiatrischen Patienten unter zehn Jahren zugelassen [siehe Anwendung bei bestimmten Patientengruppen].
BEZEICHNUNG
SEROQUEL® (Quetiapinfumarat) ist ein Psychopharmakon, das zu einer chemischen Klasse, den Dibenzothiazepin-Derivaten, gehört. Die chemische Bezeichnung lautet 2-[2-(4-Dibenzo[b,f][1,4]thiazepin-11-yl-1-piperazinyl)ethoxy]ethanolfumarat (2:1) (Salz). Es liegt in Tabletten als Fumaratsalz vor. Alle Dosierungen und Tablettenstärken sind als Milligramm der Base angegeben, nicht als Fumaratsalz. Seine Summenformel ist C42H50N6O4S2•C4H4O4 und es hat ein Molekulargewicht von 883,11 (Fumaratsalz). Die Strukturformel lautet:
Quetiapinfumarat ist ein weißes bis cremefarbenes kristallines Pulver, das in Wasser mäßig löslich ist.
SEROQUEL wird zur oralen Verabreichung als 25 mg (rund, pfirsichfarben), 50 mg (rund, weiß), 100 mg (rund, gelb), 200 mg (rund, weiß), 300 mg (kapselförmig, weiß) und geliefert 400 mg (kapselförmige, gelbe) Tabletten.
Inaktive Bestandteile sind Povidon, dibasisches Dicalciumphosphatdihydrat, mikrokristalline Cellulose, Natriumstärkeglycolat, Lactosemonohydrat, Magnesiumstearat, Hypromellose, Polyethylenglycol und Titandioxid.
Die 25-mg-Tabletten enthalten rotes Eisenoxid und gelbes Eisenoxid und die 100-mg- und 400-mg-Tabletten enthalten nur gelbes Eisenoxid.
INDIKATIONEN
Schizophrenie
SEROQUEL XR ist angezeigt zur Behandlung von Schizophrenie. Die Wirksamkeit von SEROQUEL XR bei Schizophrenie wurde in einer 6-wöchigen und einer Erhaltungsstudie bei Erwachsenen mit Schizophrenie nachgewiesen. Die Wirksamkeit wurde durch drei 6-wöchige Studien bei Erwachsenen mit Schizophrenie und eine 6-wöchige Studie bei Jugendlichen mit Schizophrenie (13-17 Jahre), die mit SEROQUEL behandelt wurden, gestützt [siehe Klinische Studien ].
Bipolare Störung
SEROQUEL 25 mg XR ist angezeigt zur akuten Behandlung von manischen oder gemischten Episoden im Zusammenhang mit einer Bipolar-I-Störung, sowohl als Monotherapie als auch als Zusatz zu Lithium oder Divalproex. Die Wirksamkeit von SEROQUEL XR bei manischen oder gemischten Episoden einer Bipolar-I-Störung wurde in einer 3-wöchigen Studie bei Erwachsenen mit manischen oder gemischten Episoden im Zusammenhang mit einer Bipolar-I-Störung nachgewiesen. Die Wirksamkeit wurde durch zwei 12-wöchige Monotherapie-Studien und eine 3-wöchige Zusatzstudie bei Erwachsenen mit manischen Episoden im Zusammenhang mit einer Bipolar-I-Störung sowie durch eine 3-wöchige Monotherapie-Studie bei Kindern und Jugendlichen (10–17 Jahre) mit manischen Episoden im Zusammenhang unterstützt mit Bipolar-I-Störung, behandelt mit SEROQUEL [siehe Klinische Studien ].
SEROQUEL 100 mg XR ist angezeigt zur akuten Behandlung depressiver Episoden im Zusammenhang mit einer bipolaren Störung. Die Wirksamkeit von SEROQUEL XR wurde in einer 8-wöchigen Studie bei Erwachsenen mit Bipolar-I- oder -II-Störung nachgewiesen und durch zwei 8-Wochen-Studien bei Erwachsenen mit Bipolar-I- oder -II-Störung, die mit SEROQUEL behandelt wurden, gestützt [siehe Klinische Studien ].
SEROQUEL 100 mg XR ist als Ergänzung zu Lithium oder Divalproex zur Erhaltungsbehandlung der Bipolar-I-Störung indiziert. Die Wirksamkeit wurde aus zwei Erhaltungsstudien bei Erwachsenen mit Bipolar-I-Störung, die mit SEROQUEL behandelt wurden, extrapoliert. Die Wirksamkeit einer Monotherapie zur Erhaltungstherapie der Bipolar-I-Störung wurde nicht systematisch in kontrollierten klinischen Studien untersucht [siehe Klinische Studien ].
Zusatzbehandlung der Major Depression (MDD)
SEROQUEL 100 mg XR ist als Begleittherapie zu Antidepressiva zur Behandlung von MDD indiziert. Die Wirksamkeit von SEROQUEL XR als Zusatztherapie zu Antidepressiva bei MDD wurde in zwei 6-wöchigen Studien bei Erwachsenen mit MDD nachgewiesen, die unzureichend auf eine Behandlung mit Antidepressiva ansprachen [siehe Klinische Studien ].
Besondere Überlegungen bei der Behandlung von pädiatrischer Schizophrenie und Bipolar-I-Störung
Pädiatrische Schizophrenie und Bipolar-I-Störung sind schwerwiegende psychische Störungen, die Diagnose kann jedoch schwierig sein. Bei der pädiatrischen Schizophrenie können die Symptomprofile variabel sein, und bei der Bipolar-I-Störung können Patienten unterschiedliche Muster der Periodizität von manischen oder gemischten Symptomen aufweisen. Es wird empfohlen, eine medikamentöse Therapie bei pädiatrischer Schizophrenie und Bipolar-I-Störung erst nach gründlicher diagnostischer Abklärung und sorgfältiger Abwägung der mit der medikamentösen Behandlung verbundenen Risiken einzuleiten. Die medikamentöse Behandlung sowohl der pädiatrischen Schizophrenie als auch der Bipolar-I-Störung ist als Teil eines Gesamtbehandlungsprogramms indiziert, das häufig psychologische, pädagogische und soziale Interventionen umfasst.
DOSIERUNG UND ANWENDUNG
Wichtige Verwaltungsanweisungen
SEROQUEL XR Tabletten sollten im Ganzen geschluckt und nicht geteilt, gekaut oder zerdrückt werden.
Es wird empfohlen, SEROQUEL 50 mg XR ohne Nahrung oder mit einer leichten Mahlzeit (ca. 300 Kalorien) einzunehmen [siehe KLINISCHE PHARMAKOLOGIE ].
SEROQUEL 25 mg XR sollte einmal täglich verabreicht werden, vorzugsweise abends.
Empfohlene Dosierung
Die empfohlene Anfangsdosis, Titration, Dosisbereich und Höchstdosis von SEROQUEL 50 mg XR für jede zugelassene Indikation sind in Tabelle 1 unten aufgeführt. Nach der Anfangsdosierung können je nach klinischem Ansprechen und Verträglichkeit des Patienten bei Bedarf Anpassungen nach oben oder unten vorgenommen werden [siehe Klinische Studien ].
Erhaltungstherapie bei Schizophrenie und Bipolar-I-Störung
Erhaltungsbehandlung
Die Patienten sollten regelmäßig neu untersucht werden, um die Notwendigkeit einer Erhaltungstherapie und die geeignete Dosis für eine solche Behandlung zu bestimmen [siehe Klinische Studien ].
Dosisanpassungen bei älteren Patienten
Bei älteren und geschwächten oder für hypotensive Reaktionen anfälligen Patienten sollte eine langsamere Dosistitration und eine niedrigere Zieldosis in Erwägung gezogen werden [siehe Verwendung in bestimmten Bevölkerungsgruppen , und KLINISCHE PHARMAKOLOGIE ]. Wenn angezeigt, sollte eine Dosissteigerung bei diesen Patienten mit Vorsicht durchgeführt werden.
Ältere Patienten sollten mit SEROQUEL XR 50 mg/Tag begonnen werden und die Dosis kann je nach klinischem Ansprechen und Verträglichkeit des einzelnen Patienten in Schritten von 50 mg/Tag erhöht werden.
Dosisänderungen bei Patienten mit eingeschränkter Leberfunktion
Patienten mit eingeschränkter Leberfunktion sollten mit SEROQUEL XR 50 mg/Tag beginnen. Die Dosis kann täglich in Schritten von 50 mg/Tag bis zu einer wirksamen Dosis erhöht werden, abhängig vom klinischen Ansprechen und der Verträglichkeit des Patienten.
Dosisänderungen bei Anwendung mit CYP3A4-Inhibitoren
Die Dosis von SEROQUEL XR sollte auf ein Sechstel der ursprünglichen Dosis reduziert werden, wenn es gleichzeitig mit einem starken CYP3A4-Hemmer (z. B. Ketoconazol, Itraconazol, Indinavir, Ritonavir, Nefazodon usw.) behandelt wird. Wenn der CYP3A4-Hemmer abgesetzt wird, sollte die Dosis von SEROQUEL XR um das 6-Fache erhöht werden [siehe KLINISCHE PHARMAKOLOGIE und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Dosisänderungen bei Verwendung mit CYP3A4-Induktoren
Die Dosis von SEROQUEL XR sollte bis auf das 5-fache der ursprünglichen Dosis erhöht werden, wenn sie in Kombination mit einer chronischen Behandlung (z. B. länger als 7-14 Tage) mit einem starken CYP3A4-Induktor (z. B. Phenytoin, Carbamazepin, Rifampin, Avasimibe, St B. Johanniskraut etc.). Die Dosis sollte basierend auf dem klinischen Ansprechen und der Verträglichkeit des einzelnen Patienten titriert werden. Wenn der CYP3A4-Induktor abgesetzt wird, sollte die Dosis von SEROQUEL 100 mg XR innerhalb von 7-14 Tagen auf das ursprüngliche Niveau reduziert werden [siehe KLINISCHE PHARMAKOLOGIE und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Wiederaufnahme der Behandlung bei Patienten, die zuvor abgebrochen wurden
Obwohl es keine Daten gibt, die sich speziell mit der Wiederaufnahme der Behandlung befassen, wird empfohlen, dass bei der Wiederaufnahme der Therapie von Patienten, die SEROQUEL 50 mg XR für mehr als eine Woche abgesetzt haben, das anfängliche Dosierungsschema befolgt werden sollte. Bei der Wiederaufnahme der Behandlung von Patienten, die SEROQUEL 300 mg XR für weniger als eine Woche abgesetzt hatten, ist möglicherweise keine schrittweise Dosissteigerung erforderlich und die Erhaltungsdosis kann wieder aufgenommen werden.
Umstellung von Patienten von SEROQUEL 50 mg Tabletten auf SEROQUEL 100 mg XR-Tabletten
Patienten, die derzeit mit SEROQUEL (Formulierung mit sofortiger Freisetzung) behandelt werden, können auf SEROQUEL XR in der äquivalenten Tagesgesamtdosis umgestellt werden, die einmal täglich eingenommen wird. Individuelle Dosisanpassungen können erforderlich sein.
Umstellung von Antipsychotika
Es liegen keine systematisch gesammelten Daten vor, die sich speziell mit der Umstellung von Patienten von anderen Antipsychotika auf SEROQUEL 200 mg XR oder mit der gleichzeitigen Verabreichung mit anderen Antipsychotika befassen. Während für einige Patienten ein sofortiges Absetzen der vorherigen antipsychotischen Behandlung akzeptabel sein kann, kann für andere ein langsameres Absetzen am besten geeignet sein. In allen Fällen sollte der Zeitraum der überlappenden Verabreichung von Antipsychotika minimiert werden. Wenn Patienten von Depot-Antipsychotika umgestellt werden, sollte, falls medizinisch angemessen, eine Behandlung mit SEROQUEL XR anstelle der nächsten planmäßigen Injektion eingeleitet werden. Die Notwendigkeit einer Fortführung der bestehenden Medikation gegen das extrapyramidale Syndrom sollte regelmäßig neu bewertet werden.
WIE GELIEFERT
Darreichungsformen und Stärken
Lagerung und Handhabung
50 mg Tabletten ( NDC 0310-0280-60) pfirsichfarbene, kapselförmige, bikonvexe Filmtablette mit Prägung „XR 50“ auf der einen Seite und glatt auf der anderen Seite sind in Flaschen mit 60 Tabletten erhältlich.
150 mg Tabletten ( NDC 0310-0281-60) weiße, filmbeschichtete, kapselförmige, bikonvexe, geprägte Tablette mit „XR 150“ auf der einen Seite und glatt auf der anderen Seite sind in Flaschen mit 60 Tabletten erhältlich.
200 mg Tabletten ( NDC 0310-0282-60) Gelbe, filmbeschichtete, kapselförmige, bikonvexe, geprägte Tablette mit „XR 200“ auf der einen Seite und glatt auf der anderen Seite sind in Flaschen mit 60 Tabletten erhältlich.
300 mg Tabletten ( NDC 0310-0283-60) blassgelbe, kapselförmige, bikonvexe Filmtablette mit Prägung „XR 300“ auf der einen Seite und glatt auf der anderen Seite sind in Flaschen mit 60 Tabletten erhältlich.
400 mg Tabletten ( NDC 0310-0284-60) weiße, filmbeschichtete, kapselförmige, bikonvexe Tablette mit Prägung „XR 400“ auf der einen Seite und glatt auf der anderen Seite sind in Flaschen mit 60 Tabletten erhältlich.
Lagern Sie SEROQUEL XR bei 25 °C (77 °F); Exkursionen erlaubt bis 15-30°C (59-86°F) [Siehe USP ].
Vertrieb durch: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. Überarbeitet: März 2020
NEBENWIRKUNGEN
Die folgenden Nebenwirkungen werden in anderen Abschnitten der Kennzeichnung ausführlicher erörtert:
Klinische Studienerfahrung
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien eines Arzneimittels beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien eines anderen Arzneimittels verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Erwachsene
Die nachstehenden Informationen stammen aus einer klinischen Studiendatenbank für SEROQUEL 200 mg, die aus über 4300 Patienten besteht. Diese Datenbank umfasst 698 Patienten, die SEROQUEL zur Behandlung von bipolarer Depression erhielten, 405 Patienten, die SEROQUEL 200 mg zur Behandlung von akuter bipolarer Manie (Monotherapie und Zusatztherapie) erhielten, 646 Patienten, die SEROQUEL zur Erhaltungsbehandlung einer Bipolar-I-Störung als Zusatztherapie erhielten Therapie, und etwa 2600 Patienten und/oder normale Probanden, die 1 oder mehr Dosen von SEROQUEL 200 mg zur Behandlung von Schizophrenie erhielten.
Von diesen etwa 4.300 Probanden waren etwa 4.000 (2.300 bei Schizophrenie, 405 bei akuter bipolarer Manie, 698 bei bipolarer Depression und 646 bei der Erhaltungstherapie einer Bipolar-I-Störung) Patienten, die an Wirksamkeitsstudien mit mehreren Dosen teilgenommen haben und deren Erfahrung entsprach ca. 2400 Patientenjahre. Die Bedingungen und Dauer der Behandlung mit SEROQUEL 200 mg waren sehr unterschiedlich und umfassten (in sich überschneidenden Kategorien) offene und doppelblinde Phasen von Studien, stationäre und ambulante Patienten, Studien mit fester Dosis und Dosistitration sowie Kurzzeit- oder Langzeitstudien Exposition. Nebenwirkungen wurden bewertet, indem Nebenwirkungen, Ergebnisse körperlicher Untersuchungen, Vitalzeichen, Gewichte, Laboranalysen, EKGs und Ergebnisse ophthalmologischer Untersuchungen gesammelt wurden.
Die angegebenen Häufigkeiten von Nebenwirkungen stellen den Anteil der Personen dar, bei denen mindestens einmal eine Nebenwirkung der aufgeführten Art aufgetreten ist.
Nebenwirkungen im Zusammenhang mit dem Absetzen der Behandlung in placebokontrollierten Kurzzeitstudien
Schizophrenie
Insgesamt gab es in einem Pool kontrollierter Studien kaum Unterschiede in der Häufigkeit von Abbrüchen aufgrund von Nebenwirkungen (4 % für SEROQUEL vs. 3 % für Placebo). Abbrüche aufgrund von Somnolenz (0,8 % SEROQUEL 100 mg vs. 0 % Placebo) und Hypotonie (0,4 % SEROQUEL 200 mg vs. 0 % Placebo) wurden jedoch als arzneimittelbedingt angesehen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Bipolare Störung
Manie
Insgesamt betrug die Abbruchrate aufgrund von Nebenwirkungen 5,7 % bei SEROQUEL 25 mg vs. 5,1 % bei Placebo in Monotherapie und 3,6 % bei SEROQUEL vs. 5,9 % bei Placebo in Zusatztherapie.
Depression
Insgesamt lagen die Abbrüche aufgrund von Nebenwirkungen bei 12,3 % bei SEROQUEL 300 mg gegenüber 19,0 % bei SEROQUEL 600 mg und 5,2 % bei Placebo.
Häufig beobachtete Nebenwirkungen in placebokontrollierten Kurzzeitstudien
In den Studien zur Akuttherapie von Schizophrenie (bis zu 6 Wochen) und bipolarer Manie (bis zu 12 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von SEROQUEL als Monotherapie (Inzidenz von 5 % oder mehr) und wurden mit einer Häufigkeit von beobachtet SEROQUEL mindestens doppelt so hoch wie Placebo waren Schläfrigkeit (18 %), Schwindel (11 %), Mundtrockenheit (9 %), Verstopfung (8 %), ALT-Erhöhung (5 %), Gewichtszunahme (5 %) und Dyspepsie ( 5 %).
Nebenwirkungen, die bei mit SEROQUEL behandelten Patienten in Placebo-kontrollierten Kurzzeitstudien mit einer Inzidenz von 2 % oder mehr auftraten
Der verschreibende Arzt sollte sich darüber im Klaren sein, dass die Zahlen in den Tabellen und Tabellen nicht dazu verwendet werden können, das Auftreten von Nebenwirkungen im Laufe der üblichen medizinischen Praxis vorherzusagen, wenn sich die Patientencharakteristika und andere Faktoren von denen unterscheiden, die in den klinischen Studien vorherrschten. Ebenso können die angegebenen Häufigkeiten nicht mit Zahlen verglichen werden, die aus anderen klinischen Studien mit unterschiedlichen Behandlungen, Anwendungen und Prüfärzten stammen. Die zitierten Zahlen bieten dem verschreibenden Arzt jedoch eine gewisse Grundlage für die Abschätzung des relativen Beitrags von medikamentösen und nichtmedikamentösen Faktoren zum Auftreten von Nebenwirkungen in der untersuchten Population.
Tabelle 9 listet die auf den nächsten Prozent gerundete Inzidenz von Nebenwirkungen auf, die während der Akuttherapie von Schizophrenie (bis zu 6 Wochen) und bipolarer Manie (bis zu 12 Wochen) bei 2 % oder mehr der mit SEROQUEL behandelten Patienten (Dosen im Bereich von von 75 auf 800 mg/Tag), wobei die Inzidenz bei mit SEROQUEL behandelten Patienten größer war als die Inzidenz bei mit Placebo behandelten Patienten.
In den Studien zur akuten Zusatztherapie der bipolaren Manie (bis zu 3 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von SEROQUEL (Inzidenz von 5 % oder mehr) und wurden unter SEROQUEL mindestens doppelt so häufig beobachtet wie unter Placebo Somnolenz (34 %), Mundtrockenheit (19 %), Asthenie (10 %), Verstopfung (10 %), Bauchschmerzen (7 %), orthostatische Hypotonie (7 %), Pharyngitis (6 %) und Gewichtszunahme (6 %).
Tabelle 10 listet die auf den nächsten Prozent gerundete Häufigkeit von Nebenwirkungen auf, die während der Therapie (bis zu 3 Wochen) der akuten Manie bei 2 % oder mehr der mit SEROQUEL (Dosen im Bereich von 100 bis 800 mg/Tag) behandelten Patienten auftraten als Zusatztherapie zu Lithium und Divalproex, wenn die Inzidenz bei mit SEROQUEL behandelten Patienten höher war als die Inzidenz bei mit Placebo behandelten Patienten.
In Studien zu bipolaren Depressionen (bis zu 8 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von SEROQUEL (Inzidenz von 5 % oder mehr) und unter SEROQUEL 200 mg mindestens doppelt so häufig wie unter Placebo beobachtete Somnolenz (57 % ), Mundtrockenheit (44 %), Schwindel (18 %), Verstopfung (10 %) und Lethargie (5 %).
Tabelle 11 führt die auf den nächsten Prozentsatz gerundete Häufigkeit von Nebenwirkungen auf, die während der Therapie (bis zu 8 Wochen) der bipolaren Depression bei 2 % oder mehr der mit SEROQUEL (Dosen von 300 und 600 mg/Tag) behandelten Patienten auftraten, wobei die Die Inzidenz bei mit SEROQUEL behandelten Patienten war höher als die Inzidenz bei mit Placebo behandelten Patienten.
Untersuchungen zu Wechselwirkungen auf der Grundlage von Geschlecht, Alter und ethnischer Zugehörigkeit ergaben keine klinisch bedeutsamen Unterschiede beim Auftreten von Nebenwirkungen auf der Grundlage dieser demografischen Faktoren.
Dosisabhängigkeit von Nebenwirkungen in Placebo-kontrollierten Kurzzeitstudien
Dosisabhängige Nebenwirkungen
Spontan erhobene Nebenwirkungen aus einer Studie über Schizophrenie, in der fünf Fixdosen von SEROQUEL (75 mg, 150 mg, 300 mg, 600 mg und 750 mg/Tag) mit Placebo verglichen wurden, wurden auf dosisabhängige Nebenwirkungen untersucht. Logistische Regressionsanalysen ergaben eine positive Dosiswirkung (p
Nebenwirkungen in klinischen Studien mit Quetiapin, die nicht an anderer Stelle auf dem Etikett aufgeführt sind:
Die folgenden Nebenwirkungen wurden auch unter Quetiapin berichtet: Albträume, Überempfindlichkeit und Erhöhungen der Kreatinphosphokinase im Serum (nicht assoziiert mit NMS), Galaktorrhoe, Bradykardie (die bei oder kurz vor Beginn der Behandlung auftreten und mit Hypotonie und/oder Synkope einhergehen können ) verringerte Blutplättchenzahl, Somnambulismus (und andere verwandte Ereignisse), Erhöhungen der Gamma-GT-Spiegel, Hypothermie, Dyspnoe, Eosinophilie, Harnverhalt, Darmverschluss und Priapismus.
Extrapyramidale Symptome (EPS)
Dystonie
Klasseneffekt
Bei empfindlichen Personen können in den ersten Tagen der Behandlung Symptome einer Dystonie, verlängerte abnorme Kontraktionen von Muskelgruppen, auftreten. Zu den dystonischen Symptomen gehören: Krämpfe der Nackenmuskulatur, die sich manchmal zu einem Engegefühl im Hals entwickeln, Schluckbeschwerden, Atembeschwerden und/oder Heraustreten der Zunge. Während diese Symptome bei niedrigen Dosen auftreten können, treten sie häufiger und mit größerer Schwere bei hochpotenten und höheren Dosen von Antipsychotika der ersten Generation auf. Ein erhöhtes Risiko einer akuten Dystonie wird bei Männern und jüngeren Altersgruppen beobachtet.
Vier Methoden wurden verwendet, um EPS zu messen: (1) Simpson-Angus-Gesamtwert (mittlere Veränderung gegenüber dem Ausgangswert), der Parkinsonismus und Akathisie bewertet, (2) Barnes Akathisia Rating Scale (BARS) Global Assessment Score, (3) Inzidenz spontaner Beschwerden von EPS (Akathisie, Akinesie, Zahnradstarrheit, extrapyramidales Syndrom, Hypertonie, Hypokinesie, Nackensteifigkeit und Tremor) und (4) Verwendung von Anticholinergika zur Behandlung von EPS.
Erwachsene
Daten aus einer 6-wöchigen klinischen Schizophrenie-Studie, in der fünf Fixdosen von SEROQUEL (75, 150, 300, 600, 750 mg/Tag) verglichen wurden, lieferten Hinweise auf das Fehlen extrapyramidaler Symptome (EPS) und die Dosisabhängigkeit von EPS im Zusammenhang mit SEROQUEL 100 mg Behandlung. Drei Methoden wurden verwendet, um EPS zu messen: (1) Simpson-Angus-Gesamtscore (mittlere Veränderung gegenüber dem Ausgangswert), der Parkinsonismus und Akathisie bewertet, (2) Inzidenz spontaner Beschwerden von EPS (Akathisie, Akinesie, Zahnradstarrheit, extrapyramidales Syndrom, Hypertonie, Hypokinesie, Nackensteifigkeit und Zittern) und (3) Verwendung von Anticholinergika gegen EPS.
In Tabelle 12 umfassten dystonische Ereignisse Nackensteifigkeit, Hypertonie, Dystonie, Muskelsteifheit, Okulogieration; Parkinsonismus umfasste Zahnradstarrheit, Zittern, Sabbern, Hypokinesie; Akathisie umfasste Akathisie, psychomotorische Erregung; dyskinetisches Ereignis umfasste tardive Dyskinesie, Dyskinesie, Choreoathetose; und andere extrapyramidale Ereignisse umfassten Ruhelosigkeit, extrapyramidale Störungen, Bewegungsstörungen.
Parkinson-Inzidenzraten, gemessen anhand des Simpson-Angus-Gesamtscores für Placebo und die fünf Fixdosen (75, 150, 300, 600, 750 mg/Tag) waren: -0,6; -1,0, -1,2; -1,6; -1,8 und -1,8. Die Rate der Verwendung von Anticholinergika zur Behandlung von EPS für Placebo und die fünf Fixdosen betrug: 14 %; 11 %; 10 %; 8 %; 12 % und 11 %.
In sechs zusätzlichen placebokontrollierten klinischen Studien (3 bei akuter Manie und 3 bei Schizophrenie) mit variablen Dosierungen von SEROQUEL gab es keine Unterschiede zwischen der SEROQUEL- und der Placebo-Behandlungsgruppe in Bezug auf das Auftreten von EPS, wie anhand der Simpson-Angus-Gesamtscores bewertet wurde. Spontanbeschwerden von EPS und die gleichzeitige Anwendung von Anticholinergika zur Behandlung von EPS.
In zwei placebokontrollierten klinischen Studien zur Behandlung von bipolarer Depression mit 300 mg und 600 mg SEROQUEL 100 mg betrug die Inzidenz von Nebenwirkungen, die möglicherweise mit EPS zusammenhängen, 12 % in beiden Dosisgruppen und 6 % in der Placebogruppe. In diesen Studien war die Inzidenz der einzelnen Nebenwirkungen (Akathisie, extrapyramidale Störung, Tremor, Dyskinesie, Dystonie, Unruhe, unwillkürliche Muskelkontraktionen, psychomotorische Hyperaktivität und Muskelrigidität) im Allgemeinen gering und überstieg in keiner Behandlungsgruppe 4 %.
Die 3 Behandlungsgruppen waren in der mittleren Veränderung des SAS-Gesamtscores und des BARS-Global-Assessment-Scores am Ende der Behandlung ähnlich. Die gleichzeitige Anwendung von Anticholinergika war selten und in allen drei Behandlungsgruppen ähnlich.
Kinder und Jugendliche
Die nachstehenden Informationen stammen aus einer klinischen Studiendatenbank für SEROQUEL 200 mg, die aus über 1000 pädiatrischen Patienten besteht. Diese Datenbank umfasst 677 Patienten, die SEROQUEL 200 mg zur Behandlung von Schizophrenie erhielten, und 393 Kinder und Jugendliche (10-17 Jahre), die SEROQUEL zur Behandlung von akuter bipolarer Manie erhielten.
Nebenwirkungen im Zusammenhang mit dem Absetzen der Behandlung in Placebo-kontrollierten Kurzzeitstudien
Schizophrenie
Die Inzidenz von Behandlungsabbrüchen aufgrund von Nebenwirkungen betrug bei mit Quetiapin behandelten und mit Placebo behandelten Patienten 8,2 % bzw. 2,7 %. Die Nebenwirkung, die bei 1 % oder mehr der Patienten unter SEROQUEL und häufiger als unter Placebo zum Absetzen führte, war Somnolenz (2,7 % und 0 % unter Placebo).
Bipolare Ich-Manie
Die Inzidenz von Behandlungsabbrüchen aufgrund von Nebenwirkungen betrug bei mit Quetiapin behandelten und mit Placebo behandelten Patienten 11,4 % bzw. 4,4 %. Die Nebenwirkungen, die bei 2 % oder mehr der Patienten unter SEROQUEL und häufiger als unter Placebo zum Absetzen führten, waren Somnolenz (4,1 % vs. 1,1 %) und Müdigkeit (2,1 % vs. 0).
Häufig beobachtete Nebenwirkungen in placebokontrollierten Kurzzeitstudien
Bei der Behandlung von Schizophrenie (bis zu 6 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von Quetiapin bei Jugendlichen (Inzidenz von 5 % oder mehr und Quetiapin-Inzidenz mindestens doppelt so hoch wie unter Placebo) Schläfrigkeit (34 %) und Schwindel (12 %), Mundtrockenheit (7 %), Tachykardie (7 %).
In der Therapie der bipolaren Manie (bis zu 3 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von Quetiapin bei Kindern und Jugendlichen (Inzidenz von 5 % oder mehr und Quetiapin-Inzidenz mindestens doppelt so hoch wie unter Placebo) Schläfrigkeit (53 %), Schwindel (18 %), Müdigkeit (11 %), gesteigerter Appetit (9 %), Übelkeit (8 %), Erbrechen (8 %), Tachykardie (7 %), Mundtrockenheit (7 %) und Gewichtszunahme (6 % ).
In einer akuten (8-wöchigen) Studie mit SEROQUEL 100 mg XR bei Kindern und Jugendlichen (10-17 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von SEROQUEL XR (Inzidenz von 5 % oder mehr und mindestens doppelt so hoch wie unter Placebo) waren Schwindel 7 %, Durchfall 5 %, Müdigkeit 5 % und Übelkeit 5 %.
Nebenwirkungen, die bei mit SEROQUEL 300 mg behandelten Patienten in Placebo-kontrollierten Kurzzeitstudien mit einer Inzidenz von ≥ 2 % auftraten
Schizophrenie (Jugendliche, 13-17 Jahre)
Die folgenden Ergebnisse basieren auf einer 6-wöchigen placebokontrollierten Studie, in der Quetiapin entweder in Dosen von 400 oder 800 mg/Tag verabreicht wurde.
Tabelle 13 führt die auf den nächsten Prozent gerundete Häufigkeit von Nebenwirkungen auf, die während der Therapie (bis zu 6 Wochen) der Schizophrenie bei 2 % oder mehr der mit SEROQUEL (Dosen von 400 oder 800 mg/Tag) behandelten Patienten auftraten, wobei die Häufigkeit angegeben ist bei mit SEROQUEL behandelten Patienten war mindestens doppelt so häufig wie bei mit Placebo behandelten Patienten.
Nebenwirkungen, die in der 800-mg-Gruppe häufiger dosisabhängig waren als in der 400-mg-Gruppe, waren Schwindel (8 % vs. 15 %), Mundtrockenheit (4 % vs. 10 %) und Tachykardie (6 % vs .11%).
Bipolar I Mania (Kinder und Jugendliche im Alter von 10-17 Jahren)
Die folgenden Ergebnisse basieren auf einer 3-wöchigen placebokontrollierten Studie, in der Quetiapin entweder in Dosen von 400 oder 600 mg/Tag verabreicht wurde.
Häufig beobachtete Nebenwirkungen
In der Therapie der bipolaren Manie (bis zu 3 Wochen) waren die am häufigsten beobachteten Nebenwirkungen im Zusammenhang mit der Anwendung von Quetiapin bei Kindern und Jugendlichen (Inzidenz von 5 % oder mehr und Quetiapin-Inzidenz mindestens doppelt so hoch wie unter Placebo) Schläfrigkeit (53 %), Schwindel (18 %), Müdigkeit (11 %), gesteigerter Appetit (9 %), Übelkeit (8 %), Erbrechen (8 %), Tachykardie (7 %), Mundtrockenheit (7 %) und Gewichtszunahme (6 % ).
Tabelle 14 führt die auf den nächsten Prozentsatz gerundete Häufigkeit von Nebenwirkungen auf, die während der Therapie (bis zu 3 Wochen) der bipolaren Manie bei 2 % oder mehr der mit SEROQUEL (Dosen von 400 oder 600 mg/Tag) behandelten Patienten auftraten, wobei die Die Inzidenz bei mit SEROQUEL behandelten Patienten war höher als die Inzidenz bei mit Placebo behandelten Patienten.
Nebenwirkungen, die in der 600-mg-Gruppe häufiger dosisabhängig waren als in der 400-mg-Gruppe, waren Schläfrigkeit (50 % vs. 57 %), Übelkeit (6 % vs. 10 %) und Tachykardie (6 % vs. 9 %).
Extrapyramidale Symptome
In einer Placebo-kontrollierten Kurzzeit-Monotherapiestudie bei jugendlichen Patienten mit Schizophrenie (Dauer 6 Wochen) betrug die aggregierte Inzidenz extrapyramidaler Symptome 12,9 % (19/147) für SEROQUEL 100 mg und 5,3 % (4/75) für Placebo. obwohl die Inzidenz der einzelnen Nebenwirkungen (Akathisie, Tremor, extrapyramidale Störung, Hypokinesie, Ruhelosigkeit, psychomotorische Hyperaktivität, Muskelrigidität, Dyskinesie) in keiner Behandlungsgruppe 4,1 % überstieg. In einer Placebo-kontrollierten Kurzzeit-Monotherapiestudie bei Kindern und Jugendlichen mit bipolarer Manie (Dauer: 3 Wochen) betrug die aggregierte Inzidenz extrapyramidaler Symptome 3,6 % (7/193) für SEROQUEL und 1,1 % (1/90) für Placebo.
Tabelle 15 enthält eine Liste von Patienten mit Nebenwirkungen, die möglicherweise mit extrapyramidalen Symptomen in der Placebo-kontrollierten Kurzzeit-Monotherapiestudie bei jugendlichen Patienten mit Schizophrenie (Dauer 6 Wochen) verbunden waren.
In den Tabellen 15–16 umfassten dystonische Ereignisse Nackensteifigkeit, Hypertonie und Muskelsteifheit; Parkinsonismus umfasste Zahnradstarrheit und Zittern; Akathisie schloss nur Akathisie ein; dyskinetisches Ereignis umfasste tardive Dyskinesie, Dyskinesie und Choreoathetose; und andere extrapyramidale Ereignisse umfassten Unruhe und extrapyramidale Störungen.
Tabelle 16 zeigt eine Liste von Patienten mit Nebenwirkungen im Zusammenhang mit extrapyramidalen Symptomen in einer Placebo-kontrollierten Kurzzeit-Monotherapiestudie bei Kindern und Jugendlichen mit bipolarer Manie (Dauer: 3 Wochen).
Labor-, EKG- und Vitalzeichenveränderungen, die in klinischen Studien beobachtet wurden
Änderungen im Labor
Anzahl der Neutrophilen
Erwachsene
In placebokontrollierten klinischen Studien zur Monotherapie mit 3368 Patienten unter Quetiapinfumarat und 1515 unter Placebo war die Inzidenz von mindestens einem Auftreten einer Neutrophilenzahl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Transaminasenerhöhungen
Erwachsene
Asymptomatische, vorübergehende und reversible Erhöhungen der Serumtransaminasen (hauptsächlich ALT) wurden berichtet. In Schizophrenie-Studien bei Erwachsenen betrug der Anteil der Patienten mit Transaminase-Erhöhungen um mehr als das Dreifache der oberen Grenzen des normalen Referenzbereichs in einem Pool von 3- bis 6-wöchigen placebokontrollierten Studien etwa 6 % (29/483) für SEROQUEL verglichen mit 1 % (3/194) bei Placebo. In Studien zur akuten bipolaren Manie bei Erwachsenen betrug der Anteil der Patienten mit Transaminase-Erhöhungen von > dem Dreifachen der Obergrenzen des normalen Referenzbereichs in einem Pool von 3- bis 12-wöchigen placebokontrollierten Studien etwa 1 % für beide SEROQUEL (3 /560) und Placebo (3/294). Diese Leberenzymerhöhungen traten normalerweise innerhalb der ersten 3 Wochen der medikamentösen Behandlung auf und kehrten bei fortlaufender Behandlung mit SEROQUEL umgehend auf die Werte vor der Studie zurück. In Studien zu bipolaren Depressionen betrug der Anteil der Patienten mit Transaminase-Erhöhungen um mehr als das Dreifache der oberen Grenzen des normalen Referenzbereichs in zwei 8-wöchigen placebokontrollierten Studien 1 % (5/698) für SEROQUEL 50 mg und 2 % (6/ 347) für Placebo.
Vermindertes Hämoglobin
Erwachsene
In placebokontrollierten Kurzzeitstudien kam es bei 8,3 % (594/7155) der mit Quetiapin behandelten Patienten mindestens einmal zu einem Abfall des Hämoglobins auf ≤ 13 g/dl bei Männern, ≤ 12 g/dl bei Frauen im Vergleich zu 6,2 % ( 219/3536) von mit Placebo behandelten Patienten. In einer Datenbank kontrollierter und unkontrollierter klinischer Studien kam es bei 11 % (2277/20729) der mit Quetiapin behandelten Patienten mindestens einmal zu einem Abfall des Hämoglobins auf ≤ 13 g/dl bei Männern und ≤ 12 g/dl bei Frauen.
Interferenz mit Urin-Drug-Screens
Es gibt Literaturberichte, die auf falsch positive Ergebnisse in Urin-Enzymimmunoassays für Methadon und trizyklische Antidepressiva bei Patienten hindeuten, die Quetiapin eingenommen haben. Bei der Interpretation positiver Urin-Drogenscreening-Ergebnisse für diese Medikamente ist Vorsicht geboten, und eine Bestätigung durch alternative Analyseverfahren (z. B. chromatographische Methoden) sollte in Betracht gezogen werden.
EKG-Veränderungen
Erwachsene
Vergleiche zwischen den Gruppen für gepoolte placebokontrollierte Studien ergaben keine statistisch signifikanten SEROQUEL/Placebo-Unterschiede in den Anteilen der Patienten, bei denen potenziell wichtige Veränderungen der EKG-Parameter, einschließlich QT-, QTc- und PR-Intervalle, auftraten. Die Anteile der Patienten, die die Kriterien für Tachykardie erfüllten, wurden jedoch in vier 3- bis 6-wöchigen placebokontrollierten klinischen Studien zur Behandlung von Schizophrenie verglichen, die eine Inzidenz von 1 % (4/399) für SEROQUEL 100 mg im Vergleich zu 0,6 % (1 /156) Inzidenz für Placebo. In Studien zu akuter (Monotherapie) bipolarer Manie betrug der Anteil der Patienten, die die Kriterien für eine Tachykardie erfüllten, 0,5 % (1/192) für SEROQUEL 200 mg im Vergleich zu 0 % (0/178) für Placebo. In Studien zur akuten bipolaren Manie (zusätzlich) betrug der Anteil der Patienten, die die gleichen Kriterien erfüllten, 0,6 % (1/166) für SEROQUEL 100 mg im Vergleich zu 0 % (0/171) für Placebo. In Studien zu bipolaren Depressionen kam es bei keinem Patienten zu einem Anstieg der Herzfrequenz auf > 120 Schläge pro Minute. Die Anwendung von SEROQUEL 50 mg war mit einem durchschnittlichen Anstieg der Herzfrequenz, bestimmt durch EKG, von 7 Schlägen pro Minute verbunden, verglichen mit einem durchschnittlichen Anstieg von 1 Schlag pro Minute bei Placebo-Patienten. Diese leichte Neigung zu Tachykardie bei Erwachsenen kann mit dem Potenzial von SEROQUEL zusammenhängen, orthostatische Veränderungen hervorzurufen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Kinder und Jugendliche
In der akuten (6-wöchigen) Schizophrenie-Studie bei Jugendlichen trat bei 5,2 % (3/73) der Patienten, die SEROQUEL 400 mg erhielten, und bei 8,5 % (5/74) der Patienten, die SEROQUEL 800 erhielten, ein Anstieg der Herzfrequenz (> 110 bpm) auf mg im Vergleich zu 0 % (0/75) der Patienten, die Placebo erhielten. Die durchschnittliche Zunahme der Herzfrequenz betrug 3,8 bpm bzw. 11,2 bpm in der Gruppe mit SEROQUEL 400 mg bzw. 800 mg, verglichen mit einer Abnahme um 3,3 bpm in der Placebogruppe [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
In der akuten (3-wöchigen) Studie zur bipolaren Manie bei Kindern und Jugendlichen trat bei 1,1 % (1/89) der Patienten, die SEROQUEL 400 mg erhielten, und bei 4,7 % (4/85) der Patienten ein Anstieg der Herzfrequenz (> 110 bpm) auf die SEROQUEL 600 mg erhielten, im Vergleich zu 0 % (0/98) der Patienten, die Placebo erhielten. Die durchschnittliche Zunahme der Herzfrequenz betrug 12,8 bpm und 13,4 bpm in der Gruppe mit SEROQUEL 400 mg bzw. 600 mg, verglichen mit einer Abnahme um 1,7 bpm in der Placebogruppe [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
In einer akuten (8-wöchigen) Studie mit SEROQUEL XR bei Kindern und Jugendlichen (10-17 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, kam es zu Anstiegen der Herzfrequenz (>110 bpm 10-12 Jahre und 13-17 Jahre) traten bei 0 % der mit SEROQUEL XR behandelten Patienten und bei 1,2 % der mit Placebo behandelten Patienten auf. Der mittlere Anstieg der Herzfrequenz betrug 3,4 bpm für SEROQUEL XR, verglichen mit 0,3 bpm in der Placebogruppe [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden nach der Zulassung von SEROQUEL identifiziert. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Seit der Markteinführung berichtete Nebenwirkungen, die in zeitlichem Zusammenhang mit der Quetiapin-Therapie standen, umfassen anaphylaktische Reaktionen, Kardiomyopathie, Arzneimittelreaktionen mit Eosinophilie und systemischen Symptomen (DRESS), Hyponatriämie, Myokarditis, nächtliches Einnässen, Pankreatitis, retrograde Amnesie, Rhabdomyolyse, Syndrom der inadäquaten ADH-Sekretion (SIADH), Stevens-Johnson-Syndrom (SJS), toxische epidermale Nekrolyse (TEN), verringerte Thrombozytenzahl, schwere Leberreaktionen (einschließlich Hepatitis, Lebernekrose und Leberversagen), Agranulozytose, Darmverschluss, Ileus, Dickdarmischämie, Harnverhalt , Schlafapnoe und akute generalisierte exanthematische Pustulose (AGEP).
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Wirkung anderer Medikamente auf Quetiapin
Die Risiken der Anwendung von SEROQUEL in Kombination mit anderen Arzneimitteln wurden nicht umfassend in systematischen Studien untersucht. Angesichts der primären ZNS-Wirkung von SEROQUEL ist Vorsicht geboten, wenn es in Kombination mit anderen zentral wirkenden Arzneimitteln eingenommen wird. SEROQUEL 300 mg potenzierte die kognitiven und motorischen Wirkungen von Alkohol in einer klinischen Studie bei Patienten mit ausgewählten psychotischen Störungen, und alkoholische Getränke sollten während der Einnahme von Quetiapin begrenzt werden.
Die Quetiapin-Exposition wird durch die Prototyp-CYP3A4-Inhibitoren (z. B. Ketoconazol, Itraconazol, Indinavir, Ritonavir, Nefazodon usw.) erhöht und durch die Prototyp-CYP3A4-Induktoren (z. B. Phenytoin, Carbamazepin, Rifampin, Avasimibe, Johanniskraut usw.) verringert. . Eine Dosisanpassung von Quetiapin ist erforderlich, wenn es gleichzeitig mit starken CYP3A4-Induktoren oder -Inhibitoren verabreicht wird.
CYP3A4-Inhibitoren
Die gleichzeitige Verabreichung von Ketoconazol, einem potenten Inhibitor des Cytochroms CYP3A4, führte zu einem signifikanten Anstieg der Quetiapin-Exposition. Die Dosis von SEROQUEL 200 mg sollte bei gleichzeitiger Anwendung mit einem starken CYP3A4-Hemmer auf ein Sechstel der ursprünglichen Dosis reduziert werden [siehe DOSIERUNG UND ANWENDUNG und KLINISCHE PHARMAKOLOGIE ].
CYP3A4-Induktoren
Die gleichzeitige Verabreichung von Quetiapin und Phenytoin, einem CYP3A4-Induktor, erhöhte die mittlere orale Clearance von Quetiapin um das Fünffache. Bei Patienten, die Quetiapin und Phenytoin oder andere bekannte potente CYP3A4-Induktoren erhalten, können erhöhte Dosen von SEROQUEL 50 mg bis zum 5-fachen erforderlich sein, um die Symptome der Schizophrenie unter Kontrolle zu halten [siehe DOSIERUNG UND ANWENDUNG und KLINISCHE PHARMAKOLOGIE ]. Wenn der CYP3A4-Induktor abgesetzt wird, sollte die Dosis von SEROQUEL 25 mg innerhalb von 7-14 Tagen auf das ursprüngliche Niveau reduziert werden [siehe DOSIERUNG UND ANWENDUNG ].
Die potenziellen Wirkungen mehrerer Begleitmedikationen auf die Pharmakokinetik von Quetiapin wurden untersucht [siehe KLINISCHE PHARMAKOLOGIE ].
Wirkung von Quetiapin auf andere Medikamente
Aufgrund seines Potenzials zur Induktion von Hypotonie kann SEROQUEL 50 mg die Wirkung bestimmter Antihypertonika verstärken.
SEROQUEL 25 mg kann die Wirkung von Levodopa und Dopaminagonisten antagonisieren.
Es gibt keine klinisch relevanten pharmakokinetischen Wechselwirkungen von Seroquel mit anderen Arzneimitteln, die auf dem CYP-Weg beruhen. Seroquel 50 mg und seine Metaboliten sind keine Inhibitoren der wichtigsten metabolisierenden CYPs (1A2, 2C9, 2C19, 2D6 und 3A4).
Drogenmissbrauch und -abhängigkeit
Kontrollierte Substanz
SEROQUEL ist keine kontrollierte Substanz.
Missbrauch
SEROQUEL wurde nicht systematisch an Tieren oder Menschen auf sein Potenzial für Missbrauch, Toleranz oder körperliche Abhängigkeit untersucht. Während die klinischen Studien keine Tendenz zu Drogensuchtverhalten aufzeigten, waren diese Beobachtungen nicht systematisch und es ist nicht möglich, auf der Grundlage dieser begrenzten Erfahrung vorherzusagen, in welchem Ausmaß ein ZNS-aktives Medikament missbraucht, abgezweigt, und/oder nach der Vermarktung missbraucht werden. Folglich sollten Patienten sorgfältig auf Drogenmissbrauch in der Vorgeschichte untersucht werden, und solche Patienten sollten engmaschig auf Anzeichen von Missbrauch oder Missbrauch von SEROQUEL beobachtet werden, z. B. Toleranzentwicklung, Dosissteigerungen, Suchtverhalten.
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose
Ältere Patienten mit demenzbedingter Psychose, die mit Antipsychotika behandelt werden, haben ein erhöhtes Sterberisiko. Die Analyse von 17 placebokontrollierten Studien (modale Dauer von 10 Wochen), hauptsächlich bei Patienten, die atypische Antipsychotika einnahmen, ergab ein Todesrisiko bei mit Arzneimitteln behandelten Patienten, das zwischen dem 1,6- und 1,7-fachen des Todesrisikos bei mit Placebo behandelten Patienten lag. Im Verlauf einer typischen 10-wöchigen kontrollierten Studie betrug die Todesrate bei mit Arzneimitteln behandelten Patienten etwa 4,5 %, verglichen mit einer Rate von etwa 2,6 % in der Placebogruppe. Obwohl die Todesursachen unterschiedlich waren, schienen die meisten Todesfälle entweder kardiovaskulärer (z. B. Herzinsuffizienz, plötzlicher Tod) oder infektiöser (z. B. Lungenentzündung) Natur zu sein. Beobachtungsstudien deuten darauf hin, dass die Behandlung mit konventionellen Antipsychotika, ähnlich wie bei atypischen Antipsychotika, die Sterblichkeit erhöhen kann. Inwieweit die Befunde einer erhöhten Sterblichkeit in Beobachtungsstudien auf das Antipsychotikum im Gegensatz zu einigen Merkmalen der Patienten zurückgeführt werden können, ist nicht klar. SEROQUEL ist nicht für die Behandlung von Patienten mit demenzbedingter Psychose zugelassen [siehe Eingerahmte Warnung ].
Suizidgedanken und -verhalten bei Jugendlichen und jungen Erwachsenen
Bei Patienten mit schwerer depressiver Störung (MDD), sowohl bei Erwachsenen als auch bei Kindern, kann es zu einer Verschlechterung ihrer Depression und/oder zum Auftreten von Suizidgedanken und -verhalten (Suizidalität) oder zu ungewöhnlichen Verhaltensänderungen kommen, unabhängig davon, ob sie Antidepressiva einnehmen oder nicht Das Risiko kann bestehen bleiben, bis eine signifikante Remission eintritt. Selbstmord ist ein bekanntes Risiko für Depressionen und bestimmte andere psychiatrische Störungen, und diese Störungen selbst sind die stärksten Prädiktoren für Selbstmord. Es besteht jedoch seit langem die Sorge, dass Antidepressiva bei bestimmten Patienten in den frühen Phasen der Behandlung eine Rolle bei der Induktion einer Verschlechterung der Depression und dem Auftreten von Suizidalität spielen könnten. Gepoolte Analysen von Placebo-kontrollierten Kurzzeitstudien mit Antidepressiva (SSRIs und andere) zeigten, dass diese Medikamente das Risiko von Suizidgedanken und -verhalten (Suizidalität) bei Kindern, Jugendlichen und jungen Erwachsenen (18-24 Jahre) mit Major Depression erhöhen Störung (MDD) und andere psychiatrische Störungen. Kurzzeitstudien zeigten bei Erwachsenen über 24 Jahren keine Erhöhung des Suizidalitätsrisikos mit Antidepressiva im Vergleich zu Placebo; bei Erwachsenen ab 65 Jahren kam es unter Antidepressiva im Vergleich zu Placebo zu einer Reduktion.
Die gepoolten Analysen placebokontrollierter Studien bei Kindern und Jugendlichen mit MDD, Zwangsstörungen (OCD) oder anderen psychiatrischen Erkrankungen umfassten insgesamt 24 Kurzzeitstudien mit 9 Antidepressiva bei über 4400 Patienten. Die gepoolten Analysen placebokontrollierter Studien bei Erwachsenen mit MDD oder anderen psychiatrischen Erkrankungen umfassten insgesamt 295 Kurzzeitstudien (mediane Dauer von 2 Monaten) mit 11 Antidepressiva bei über 77.000 Patienten. Es gab beträchtliche Schwankungen des Suizidalitätsrisikos zwischen den Medikamenten, aber bei fast allen untersuchten Medikamenten eine Tendenz zu einem Anstieg bei den jüngeren Patienten. Es gab Unterschiede im absoluten Suizidalitätsrisiko zwischen den verschiedenen Indikationen, mit der höchsten Inzidenz bei MDD. Die Risikounterschiede (Medikament vs. Placebo) waren jedoch relativ stabil innerhalb der Altersschichten und über Indikationen hinweg. Diese Risikounterschiede (Arzneimittel-Placebo-Unterschied in der Anzahl der Fälle von Suizidalität pro 1000 behandelten Patienten) sind in Tabelle 2 aufgeführt.
In keiner der pädiatrischen Studien kam es zu Suiziden. In den Studien mit Erwachsenen gab es Suizide, aber die Anzahl reichte nicht aus, um zu einer Schlussfolgerung über die Wirkung des Medikaments auf Suizide zu gelangen.
Es ist nicht bekannt, ob sich das Suizidrisiko auf eine längerfristige Anwendung erstreckt, dh über mehrere Monate hinaus. Es gibt jedoch erhebliche Hinweise aus placebokontrollierten Erhaltungsstudien bei Erwachsenen mit Depressionen, dass die Anwendung von Antidepressiva das Wiederauftreten von Depressionen verzögern kann.
Alle Patienten, die mit Antidepressiva aus beliebigen Indikationen behandelt werden, sollten angemessen überwacht und engmaschig auf eine klinische Verschlechterung, Suizidalität und ungewöhnliche Verhaltensänderungen beobachtet werden, insbesondere während der ersten Monate einer medikamentösen Therapie oder bei Dosisänderungen oder -steigerungen oder abnimmt.
Die folgenden Symptome, Angst, Agitiertheit, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Ruhelosigkeit), Hypomanie und Manie, wurden bei erwachsenen und pädiatrischen Patienten berichtet, die ebenfalls mit Antidepressiva wegen schwerer depressiver Störung behandelt wurden wie für andere Indikationen, sowohl psychiatrische als auch nicht-psychiatrische. Obwohl ein kausaler Zusammenhang zwischen dem Auftreten solcher Symptome und entweder der Verschlechterung einer Depression und/oder dem Auftreten suizidaler Impulse nicht hergestellt werden konnte, besteht die Sorge, dass solche Symptome Vorboten einer aufkommenden Suizidalität sein könnten.
Bei Patienten, deren Depression sich anhaltend verschlimmert oder die an Suizidalität oder Symptomen leiden, die Vorboten einer Verschlechterung der Depression oder Suizidalität sein könnten, sollte eine Änderung des therapeutischen Schemas, einschließlich eines möglichen Absetzens der Medikation, in Betracht gezogen werden, insbesondere wenn diese Symptome schwerwiegend und abrupt sind zu Beginn oder waren nicht Teil der Symptome des Patienten.
Familien und Betreuer von Patienten, die wegen einer schweren depressiven Störung oder anderen psychiatrischen und nicht-psychiatrischen Indikationen mit Antidepressiva behandelt werden, sollten auf die Notwendigkeit aufmerksam gemacht werden, die Patienten auf das Auftreten von Unruhe, Reizbarkeit, ungewöhnlichen Verhaltensänderungen und anderen Symptomen zu überwachen wie oben beschrieben, sowie das Auftreten von Suizidalität, und solche Symptome unverzüglich dem Gesundheitsdienstleister zu melden. Eine solche Überwachung sollte die tägliche Beobachtung durch Familien und Betreuer umfassen. Verschreibungen für SEROQUEL sollten für die kleinste Tablettenmenge im Einklang mit einem guten Patientenmanagement ausgestellt werden, um das Risiko einer Überdosierung zu verringern.
Screening von Patienten auf bipolare Störungen
Eine schwere depressive Episode kann die anfängliche Präsentation einer bipolaren Störung sein. Es wird allgemein angenommen (obwohl dies nicht in kontrollierten Studien nachgewiesen wurde), dass die Behandlung einer solchen Episode mit einem Antidepressivum allein die Wahrscheinlichkeit des Auftretens einer gemischten/manischen Episode bei Patienten mit einem Risiko für eine bipolare Störung erhöhen kann. Ob eines der oben beschriebenen Symptome eine solche Konversion darstellt, ist unbekannt. Vor Beginn der Behandlung mit einem Antidepressivum, einschließlich SEROQUEL, sollten Patienten mit depressiven Symptomen jedoch angemessen untersucht werden, um festzustellen, ob bei ihnen ein Risiko für eine bipolare Störung besteht; Ein solches Screening sollte eine detaillierte psychiatrische Vorgeschichte umfassen, einschließlich einer Familienanamnese von Selbstmord, bipolarer Störung und Depression.
Zerebrovaskuläre Nebenwirkungen, einschließlich Schlaganfall, bei älteren Patienten mit demenzbedingter Psychose
In placebokontrollierten Studien mit Risperidon, Aripiprazol und Olanzapin bei älteren Patienten mit Demenz traten im Vergleich zu den mit Placebo behandelten Patienten häufiger zerebrovaskuläre Nebenwirkungen (zerebrovaskuläre Insultate und transitorische ischämische Attacken) auf, einschließlich Todesfälle. SEROQUEL ist nicht für die Behandlung von Patienten mit demenzbedingter Psychose zugelassen [siehe auch Eingerahmte Warnung und Erhöhte Sterblichkeit bei älteren Patienten mit demenzbedingter Psychose ].
Malignes neuroleptisches Syndrom (NMS)
Im Zusammenhang mit der Verabreichung von Antipsychotika, einschließlich SEROQUEL, wurde über einen potenziell tödlichen Symptomkomplex berichtet, der manchmal als malignes neuroleptisches Syndrom (NMS) bezeichnet wird. Unter SEROQUEL wurden seltene Fälle von NMS berichtet. Klinische Manifestationen von NMS sind Hyperpyrexie, Muskelrigidität, veränderter Geisteszustand und Anzeichen einer autonomen Instabilität (unregelmäßiger Puls oder Blutdruck, Tachykardie, Diaphorese und Herzrhythmusstörungen). Zusätzliche Anzeichen können erhöhte Kreatininphosphokinase, Myoglobinurie (Rhabdomyolyse) und akutes Nierenversagen sein.
Die diagnostische Beurteilung von Patienten mit diesem Syndrom ist kompliziert. Um zu einer Diagnose zu gelangen, ist es wichtig, Fälle auszuschließen, in denen das klinische Erscheinungsbild sowohl eine schwere medizinische Erkrankung (z. B. Lungenentzündung, systemische Infektion usw.) als auch unbehandelte oder unzureichend behandelte extrapyramidale Zeichen und Symptome (EPS) umfasst. Weitere wichtige Erwägungen bei der Differentialdiagnose sind zentrale anticholinerge Toxizität, Hitzschlag, Drogenfieber und primäre Pathologie des Zentralnervensystems (ZNS).
Das Management von NMS sollte umfassen: 1) sofortiges Absetzen von Antipsychotika und anderen Arzneimitteln, die für eine gleichzeitige Therapie nicht unbedingt erforderlich sind; 2) intensive symptomatische Behandlung und medizinische Überwachung; und 3) Behandlung aller begleitenden schwerwiegenden medizinischen Probleme, für die spezifische Behandlungen verfügbar sind. Es gibt keine allgemeine Einigung über spezifische pharmakologische Behandlungsschemata für NMS.
Wenn ein Patient nach Genesung von NMS eine antipsychotische medikamentöse Behandlung benötigt, sollte die mögliche Wiederaufnahme der medikamentösen Therapie sorgfältig erwogen werden. Der Patient sollte sorgfältig überwacht werden, da Rezidive von NMS berichtet wurden.
Stoffwechselveränderungen
Atypische Antipsychotika wurden mit metabolischen Veränderungen wie Hyperglykämie/Diabetes mellitus, Dyslipidämie und Körpergewichtszunahme in Verbindung gebracht. Obwohl gezeigt wurde, dass alle Medikamente dieser Klasse einige metabolische Veränderungen hervorrufen, hat jedes Medikament sein eigenes spezifisches Risikoprofil. Bei einigen Patienten wurde in klinischen Studien eine Verschlechterung von mehr als einem der Stoffwechselparameter Gewicht, Blutzucker und Lipide beobachtet. Änderungen dieser Stoffwechselprofile sollten klinisch angemessen behandelt werden.
Hyperglykämie und Diabetes mellitus
Hyperglykämie, in einigen Fällen extrem und mit Ketoazidose oder hyperosmolarem Koma oder Tod verbunden, wurde bei Patienten berichtet, die mit atypischen Antipsychotika, einschließlich Quetiapin, behandelt wurden. Die Beurteilung des Zusammenhangs zwischen der atypischen Anwendung von Antipsychotika und Glukoseanomalien wird durch die Möglichkeit eines erhöhten Hintergrundrisikos für Diabetes mellitus bei Patienten mit Schizophrenie und die zunehmende Inzidenz von Diabetes mellitus in der Allgemeinbevölkerung erschwert. Angesichts dieser Confounder ist die Beziehung zwischen der atypischen Anwendung von Antipsychotika und Hyperglykämie-bedingten Nebenwirkungen nicht vollständig geklärt. Epidemiologische Studien deuten jedoch auf ein erhöhtes Risiko für Hyperglykämie-bedingte Nebenwirkungen bei Patienten hin, die mit atypischen Antipsychotika behandelt werden. Präzise Risikoabschätzungen für Hyperglykämie-bedingte Nebenwirkungen bei Patienten, die mit atypischen Antipsychotika behandelt werden, sind nicht verfügbar.
Patienten mit einer gesicherten Diagnose von Diabetes mellitus, die mit atypischen Antipsychotika begonnen werden, sollten regelmäßig auf eine Verschlechterung der Glukosekontrolle überwacht werden. Patienten mit Risikofaktoren für Diabetes mellitus (z. B. Fettleibigkeit, Diabetes in der Familienanamnese), die eine Behandlung mit atypischen Antipsychotika beginnen, sollten zu Beginn der Behandlung und in regelmäßigen Abständen während der Behandlung einer Nüchtern-Blutzuckermessung unterzogen werden. Jeder Patient, der mit atypischen Antipsychotika behandelt wird, sollte auf Symptome einer Hyperglykämie einschließlich Polydipsie, Polyurie, Polyphagie und Schwäche überwacht werden. Patienten, die während der Behandlung mit atypischen Antipsychotika Symptome einer Hyperglykämie entwickeln, sollten sich einer Nüchtern-Blutzuckermessung unterziehen. In einigen Fällen klang die Hyperglykämie ab, wenn das atypische Antipsychotikum abgesetzt wurde; Einige Patienten benötigten jedoch trotz Absetzens des verdächtigen Medikaments eine Fortsetzung der antidiabetischen Behandlung.
Erwachsene
In einer 24-wöchigen Studie (aktiv kontrolliert, 115 mit SEROQUEL behandelte Patienten), die darauf ausgelegt war, den glykämischen Status mit oralen Glukosetoleranztests bei allen Patienten zu bewerten, betrug die Inzidenz eines Glukosespiegels nach der Glukosebelastung in Woche 24 ≥ 200 mg/dl 1,7 % und die Inzidenz eines Nüchtern-Blutzuckerspiegels ≥ 126 mg/dl betrug 2,6 %. Die mittlere Veränderung des Nüchternglukosespiegels gegenüber dem Ausgangswert betrug 3,2 mg/dl und die mittlere Veränderung des 2-Stunden-Glukosespiegels gegenüber dem Ausgangswert betrug -1,8 mg/dl für Quetiapin.
In 2 placebokontrollierten, randomisierten klinischen Langzeit-Entzugsstudien zur Aufrechterhaltung der Bipolar-I-Störung, mittlere Exposition von 213 Tagen für SEROQUEL (646 Patienten) und 152 Tage für Placebo (680 Patienten), betrug die mittlere Veränderung des Glukosespiegels gegenüber dem Ausgangswert +5,0 mg /dL für SEROQUEL 100 mg und –0,05 mg/dL für Placebo. Die expositionsbereinigte Rate für jeden erhöhten Blutzuckerspiegel (≥ 126 mg/dl) bei Patienten, die mehr als 8 Stunden nach einer Mahlzeit vergangen waren (einige Patienten wurden jedoch möglicherweise nicht von der Kalorienaufnahme durch Flüssigkeiten während der Fastenzeit ausgeschlossen) betrug 18,0 pro 100 Patientenjahre für SEROQUEL (10,7 % der Patienten; n = 556) und 9,5 für Placebo pro 100 Patientenjahre (4,6 % der Patienten; n = 581).
Kinder und Jugendliche
In einer placebokontrollierten Monotherapiestudie mit SEROQUEL 25 mg an jugendlichen Patienten (13-17 Jahre) mit Schizophrenie (Dauer 6 Wochen) war die mittlere Veränderung der Nüchternglukosewerte für SEROQUEL (n = 138) im Vergleich zu Placebo (n = 67) betrug – 0,75 mg/dL gegenüber –1,70 mg/dL. In einer placebokontrollierten Monotherapiestudie mit SEROQUEL 300 mg bei Kindern und Jugendlichen (10–17 Jahre) mit bipolarer Manie (Dauer: 3 Wochen) war die mittlere Veränderung des Nüchternglukosespiegels für SEROQUEL (n = 170) im Vergleich zu Placebo (n =81) betrug 3,62 mg/dl gegenüber –1,17 mg/dl. Kein Patient in beiden Studien mit einem normalen Nüchternglukosespiegel (
In einer placebokontrollierten SEROQUEL XR-Monotherapiestudie (Dauer 8 Wochen) mit Kindern und jugendlichen Patienten (1017 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, war die mittlere Veränderung des Nüchternglukosespiegels für SEROQUEL 300 mg XR (n= 60) im Vergleich zu Placebo (n=62) betrug 1,8 mg/dl gegenüber 1,6 mg/dl. In dieser Studie gab es keine Patienten in den mit SEROQUEL XR oder Placebo behandelten Gruppen mit einem normalen Nüchternglukosespiegel ( 126 mg/dl aufwiesen. Es gab einen Patienten in der SEROQUEL 25 mg XR-Gruppe mit einem grenzwertigen Nüchternglukosewert (> 100 mg/dl und 126 mg/dl im Vergleich zu null Patienten in der Gruppe aufwies Placebo-Gruppe.
Dyslipidämie
Erwachsene
Tabelle 4 zeigt den Prozentsatz erwachsener Patienten mit Veränderungen des Gesamtcholesterins, der Triglyceride, des LDL-Cholesterins und des HDL-Cholesterins gegenüber dem Ausgangswert nach Indikation in klinischen Studien mit SEROQUEL.
Kinder und Jugendliche
Tabelle 5 zeigt den prozentualen Anteil von Kindern und Jugendlichen mit Veränderungen des Gesamtcholesterins, der Triglyzeride, des LDL-Cholesterins und des HDL-Cholesterins gegenüber dem Ausgangswert in klinischen Studien mit SEROQUEL.
In einer placebokontrollierten SEROQUEL XR-Monotherapiestudie (Dauer 8 Wochen) mit Kindern und Jugendlichen (1017 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, war der Prozentsatz der Kinder und Jugendlichen mit Verschiebungen des Gesamtcholesterins (≥200 mg/dl), Triglyceride (≥ 150 mg/dl), LDL-Cholesterin (≥ 130 mg/dl) und HDL-Cholesterin (≤ 40 mg/dl) vom Ausgangswert bis zu klinisch signifikanten Werten: Gesamtcholesterin 8 % (7 /83) für SEROQUEL 50 mg XR vs. 6 % (5/84) für Placebo; Triglyceride 28 % (22/80) für SEROQUEL 100 mg XR vs. 9 % (7/82) für Placebo; LDL-Cholesterin 2 % (2/86) für SEROQUEL XR vs. 4 % (3/85) für Placebo und HDL-Cholesterin 20 % (13/65) für SEROQUEL XR vs. 15 % (11/74) für Placebo.
Gewichtszunahme
In klinischen Studien wurde eine Gewichtszunahme beobachtet. Patienten, die Quetiapin erhalten, sollten regelmäßig ihr Gewicht kontrollieren.
Erwachsene
In klinischen Studien mit SEROQUEL 50 mg wurde über folgende Gewichtszunahmen berichtet.
Kinder und Jugendliche
In zwei klinischen Studien mit SEROQUEL 25 mg, eine bei bipolarer Manie und eine bei Schizophrenie, sind die berichteten Gewichtszunahmen in Tabelle 7 enthalten.
Die mittlere Veränderung des Körpergewichts betrug in der Schizophrenie-Studie 2,0 kg in der SEROQUEL-25-mg-Gruppe und -0,4 kg in der Placebo-Gruppe und in der Bipolar-Mania-Studie 1,7 kg in der SEROQUEL-200-mg-Gruppe und 0,4 kg in der Placebo-Gruppe.
In einer offenen Studie, in die Patienten aus den beiden oben genannten pädiatrischen Studien aufgenommen wurden, beendeten 63 % der Patienten (241/380) eine 26-wöchige Therapie mit SEROQUEL. Nach 26-wöchiger Behandlung betrug die mittlere Zunahme des Körpergewichts 4,4 kg. 45 % der Patienten nahmen ≥ 7 % ihres Körpergewichts zu, nicht angepasst an normales Wachstum. Um ein normales Wachstum über 26 Wochen auszugleichen, wurde eine Erhöhung des BMI um mindestens 0,5 Standardabweichung vom Ausgangswert als Maß für eine klinisch signifikante Veränderung verwendet; 18,3 % der mit SEROQUEL behandelten Patienten erfüllten dieses Kriterium nach 26 Behandlungswochen.
In einer klinischen Studie mit SEROQUEL XR bei Kindern und Jugendlichen (10-17 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, betrug der Prozentsatz der Patienten mit einer Gewichtszunahme von ≥ 7 % des Körpergewichts zu irgendeinem Zeitpunkt 15 % ( 14/92) für SEROQUEL 25 mg XR gegenüber 10 % (10/100) für Placebo. Die mittlere Veränderung des Körpergewichts betrug 1,4 kg in der Gruppe mit SEROQUEL 50 mg XR gegenüber 0,6 kg in der Placebogruppe.
Bei der Behandlung von pädiatrischen Patienten mit SEROQUEL 50 mg für jede Indikation sollte die Gewichtszunahme im Vergleich zu der erwarteten Gewichtszunahme bei normalem Wachstum beurteilt werden.
Tardive Dyskinesie
Bei Patienten, die mit Antipsychotika, einschließlich Quetiapin, behandelt werden, kann sich ein Syndrom potenziell irreversibler, unwillkürlicher, dyskinetischer Bewegungen entwickeln. Obwohl die Prävalenz des Syndroms bei älteren Menschen, insbesondere bei älteren Frauen, am höchsten zu sein scheint, ist es unmöglich, sich auf Prävalenzschätzungen zu verlassen, um zu Beginn einer antipsychotischen Behandlung vorherzusagen, welche Patienten wahrscheinlich das Syndrom entwickeln werden. Ob Antipsychotika sich in ihrem Potenzial unterscheiden, tardive Dyskinesie zu verursachen, ist nicht bekannt.
Es wird angenommen, dass das Risiko der Entwicklung einer tardiven Dyskinesie und die Wahrscheinlichkeit, dass sie irreversibel wird, mit der Dauer der Behandlung und der kumulativen Gesamtdosis der dem Patienten verabreichten Antipsychotika zunimmt. Das Syndrom kann sich jedoch, wenn auch viel seltener, nach relativ kurzen Behandlungszeiten bei niedrigen Dosen entwickeln oder sogar nach Absetzen der Behandlung auftreten.
Spätdyskinesie kann teilweise oder vollständig zurückgehen, wenn die antipsychotische Behandlung abgesetzt wird. Die antipsychotische Behandlung selbst kann jedoch die Anzeichen und Symptome des Syndroms unterdrücken (oder teilweise unterdrücken) und dadurch möglicherweise den zugrunde liegenden Prozess maskieren. Die Auswirkung der symptomatischen Unterdrückung auf den Langzeitverlauf des Syndroms ist unbekannt.
Angesichts dieser Überlegungen sollte SEROQUEL 50 mg so verschrieben werden, dass das Auftreten von tardiven Dyskinesien am wahrscheinlichsten minimiert wird. Eine chronische Behandlung mit Antipsychotika sollte im Allgemeinen Patienten vorbehalten bleiben, die anscheinend an einer chronischen Krankheit leiden, die (1) bekanntermaßen auf Antipsychotika anspricht und (2) für die alternative, ebenso wirksame, aber potenziell weniger schädliche Behandlungen nicht verfügbar oder angemessen sind . Bei Patienten, die eine chronische Behandlung benötigen, sollte die niedrigste Dosis und die kürzeste Behandlungsdauer angestrebt werden, die ein zufriedenstellendes klinisches Ansprechen bewirken. Die Notwendigkeit einer Fortsetzung der Behandlung sollte regelmäßig überprüft werden.
Wenn bei einem Patienten unter SEROQUEL 200 mg Anzeichen und Symptome einer tardiven Dyskinesie auftreten, sollte ein Absetzen des Arzneimittels in Erwägung gezogen werden. Einige Patienten können jedoch trotz des Vorliegens des Syndroms eine Behandlung mit SEROQUEL 200 mg benötigen.
Hypotonie
Quetiapin kann eine orthostatische Hypotonie hervorrufen, die mit Schwindel, Tachykardie und bei manchen Patienten mit Synkopen einhergeht, insbesondere während der Anfangsphase der Dosistitration, was wahrscheinlich seine α1-adrenergen antagonistischen Eigenschaften widerspiegelt. Synkopen wurden bei 1 % (28/3265) der mit SEROQUEL behandelten Patienten berichtet, verglichen mit 0,2 % (2/954) unter Placebo und etwa 0,4 % (2/527) unter aktiven Kontrollmedikamenten. Orthostatische Hypotonie, Schwindel und Synkopen können zu Stürzen führen.
SEROQUEL 25 mg sollte bei Patienten mit bekannter kardiovaskulärer Erkrankung (Myokardinfarkt oder ischämische Herzerkrankung in der Vorgeschichte, Herzinsuffizienz oder Reizleitungsstörungen), zerebrovaskulärer Erkrankung oder Zuständen, die Patienten für Hypotonie prädisponieren würden (Dehydratation, Hypovolämie und Behandlung mit blutdrucksenkende Medikamente). Das Risiko einer orthostatischen Hypotonie und Synkope kann minimiert werden, indem die Anfangsdosis auf 25 mg zweimal täglich begrenzt wird [siehe DOSIERUNG UND ANWENDUNG ]. Wenn während der Titration auf die Zieldosis eine Hypotonie auftritt, ist eine Rückkehr zur vorherigen Dosis im Titrationsplan angemessen.
Stürze
Atypische Antipsychotika, einschließlich SEROQUEL, können Somnolenz, posturale Hypotonie, motorische und sensorische Instabilität verursachen, was zu Stürzen und folglich zu Knochenbrüchen oder anderen Verletzungen führen kann. Führen Sie bei Patienten mit Krankheiten, Zuständen oder Medikamenten, die diese Wirkungen verschlimmern könnten, zu Beginn einer antipsychotischen Behandlung und wiederholt bei Patienten unter antipsychotischer Langzeittherapie eine Sturzrisikobewertung durch.
Anstieg des Blutdrucks (Kinder und Jugendliche)
In placebokontrollierten Studien bei Kindern und Jugendlichen mit Schizophrenie (Dauer 6 Wochen) oder bipolarer Manie (Dauer 3 Wochen) betrug die Inzidenz eines Anstiegs des systolischen Blutdrucks (≥ 20 mmHg) zu irgendeinem Zeitpunkt 15,2 % (51/335 ) für SEROQUEL 50 mg und 5,5 % (9/163) für Placebo; die Häufigkeit von Erhöhungen des diastolischen Blutdrucks (≥ 10 mmHg) zu irgendeinem Zeitpunkt betrug 40,6 % (136/335) für SEROQUEL und 24,5 % (40/163) für Placebo. In der 26-wöchigen offenen klinischen Studie erlitt ein Kind mit bekannter Vorgeschichte von Bluthochdruck eine hypertensive Krise. Der Blutdruck bei Kindern und Jugendlichen sollte zu Beginn und in regelmäßigen Abständen während der Behandlung gemessen werden.
In einer placebokontrollierten klinischen Studie mit SEROQUEL XR (Dauer 8 Wochen) bei Kindern und Jugendlichen (10-17 Jahre) mit bipolarer Depression, in der die Wirksamkeit nicht nachgewiesen wurde, war die Inzidenz von Anstiegen des systolischen Blutdrucks (≥ 20 mmHg) betrug 6,5 % (6/92) für SEROQUEL XR und 6,0 % (6/100) für Placebo; die Häufigkeit von Erhöhungen des diastolischen Blutdrucks (≥ 10 mmHg) zu irgendeinem Zeitpunkt betrug 46,7 % (43/92) für SEROQUEL 25 mg XR und 36,0 % (36/100) für Placebo.
Leukopenie, Neutropenie und Agranulozytose
In klinischen Studien und nach Markteinführung wurden Fälle von Leukopenie/Neutropenie in zeitlichem Zusammenhang mit atypischen Antipsychotika, einschließlich SEROQUEL, berichtet. Agranulozytose wurde berichtet.
Unter Quetiapin wurde über Agranulozytose (definiert als absolute Neutrophilenzahl
Mögliche Risikofaktoren für Leukopenie/Neutropenie sind eine vorbestehende niedrige Anzahl weißer Blutkörperchen (WBC) und eine arzneimittelinduzierte Leukopenie/Neutropenie in der Vorgeschichte. Bei Patienten mit vorbestehender niedriger Leukozytenzahl oder arzneimittelinduzierter Leukopenie/Neutropenie in der Vorgeschichte sollte ihr komplettes Blutbild (CBC) während der ersten Monate der Therapie häufig überwacht werden und SEROQUEL 50 mg beim ersten Anzeichen einer Abnahme der Leukozytenzahl abgesetzt werden Fehlen anderer ursächlicher Faktoren.
Patienten mit Neutropenie sollten sorgfältig auf Fieber oder andere Symptome oder Anzeichen einer Infektion überwacht und unverzüglich behandelt werden, wenn solche Symptome oder Anzeichen auftreten. Patienten mit schwerer Neutropenie (absolute Neutrophilenzahl
Grauer Star
Die Entwicklung von Katarakten wurde im Zusammenhang mit der Behandlung mit Quetiapin in Studien mit chronischen Hunden beobachtet [vgl Nichtklinische Toxikologie ]. Linsenveränderungen wurden auch bei Erwachsenen, Kindern und Jugendlichen während der Langzeitbehandlung mit SEROQUEL 100 mg beobachtet, aber ein kausaler Zusammenhang mit der Anwendung von SEROQUEL 200 mg wurde nicht hergestellt. Dennoch kann die Möglichkeit von Linsenveränderungen zum jetzigen Zeitpunkt nicht ausgeschlossen werden. Daher wird zu Beginn der Behandlung oder kurz danach und während einer Dauerbehandlung in Abständen von 6 Monaten eine Untersuchung der Linse mit Methoden empfohlen, die zum Nachweis einer Kataraktbildung geeignet sind, wie z. B. eine Spaltlampenuntersuchung oder andere angemessen empfindliche Methoden.
QT-Verlängerung
In klinischen Studien war Quetiapin nicht mit einer anhaltenden Verlängerung der QT-Intervalle verbunden. Der QT-Effekt wurde jedoch nicht systematisch in einer gründlichen QT-Studie evaluiert. Nach der Markteinführung wurden Fälle von QT-Verlängerung bei Patienten berichtet, die Quetiapin überdosiert hatten [siehe ÜBERDOSIS ], bei Patienten mit Begleiterkrankungen und bei Patienten, die Arzneimittel einnehmen, die bekanntermaßen ein Elektrolytungleichgewicht verursachen oder das QT-Intervall verlängern [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Die Anwendung von Quetiapin sollte in Kombination mit anderen Arzneimitteln vermieden werden, die bekanntermaßen das QTc verlängern, einschließlich Antiarrhythmika der Klasse 1A (z. B. Chinidin, Procainamid) oder Antiarrhythmika der Klasse III (z. B. Amiodaron, Sotalol), Antipsychotika (z. B. Ziprasidon, Chlorpromazin, Thioridazin), Antibiotika (z. B. Gatifloxacin, Moxifloxacin) oder jede andere Klasse von Medikamenten, von denen bekannt ist, dass sie das QTc-Intervall verlängern (z. B. Pentamidin, Levomethadylacetat, Methadon).
Quetiapin sollte auch unter Umständen vermieden werden, die das Risiko des Auftretens von Torsade de Pointes und/oder plötzlichem Tod erhöhen können, einschließlich (1) einer Vorgeschichte von Herzrhythmusstörungen wie Bradykardie; (2) Hypokaliämie oder Hypomagnesiämie; (3) gleichzeitige Anwendung anderer Arzneimittel, die das QTc-Intervall verlängern; und (4) Vorhandensein einer angeborenen Verlängerung des QT-Intervalls.
Vorsicht ist auch geboten, wenn Quetiapin Patienten mit erhöhtem Risiko einer QT-Verlängerung verschrieben wird (z. B. Herz-Kreislauf-Erkrankungen, QT-Verlängerung in der Familienanamnese, ältere Menschen, dekompensierte Herzinsuffizienz und Herzhypertrophie).
Krampfanfälle
Während klinischer Studien traten bei 0,5 % (20/3490) der mit SEROQUEL behandelten Patienten Krampfanfälle auf, verglichen mit 0,2 % (2/954) unter Placebo und 0,7 % (4/527) unter aktiven Kontrollmedikamenten. Wie andere Antipsychotika sollte SEROQUEL 25 mg bei Patienten mit Krampfanfällen in der Vorgeschichte oder mit Erkrankungen, die möglicherweise die Krampfschwelle senken, z. B. Alzheimer-Demenz, mit Vorsicht angewendet werden. Zustände, die die Anfallsschwelle senken, können in einer Bevölkerung von 65 Jahren oder älter häufiger auftreten.
Hypothyreose
Erwachsene
Klinische Studien mit Quetiapin zeigten dosisabhängige Abnahmen der Schilddrüsenhormonspiegel. Die Reduktion des Gesamt- und freien Thyroxins (T4) um etwa 20 % am oberen Ende des therapeutischen Dosisbereichs war in den ersten sechs Behandlungswochen maximal und blieb ohne Anpassung oder Progression während einer chronischeren Therapie bestehen. In fast allen Fällen war das Absetzen der Behandlung mit Quetiapin unabhängig von der Behandlungsdauer mit einer Umkehrung der Wirkungen auf Gesamt- und freies T4 verbunden. Der Mechanismus, durch den Quetiapin die Schilddrüsenachse beeinflusst, ist unklar. Wenn es eine Auswirkung auf die Hypothalamus-Hypophysen-Achse gibt, spiegelt die Messung von TSH allein möglicherweise nicht genau den Schilddrüsenstatus eines Patienten wider. Daher sollten sowohl TSH als auch freies T4 zusätzlich zur klinischen Beurteilung zu Studienbeginn und bei der Nachsorge gemessen werden.
In den Begleitstudien zur Manie, in denen SEROQUEL 50 mg zu Lithium oder Divalproex hinzugefügt wurde, hatten 12 % (24/196) der mit SEROQUEL 200 mg behandelten Patienten im Vergleich zu 7 % (15/203) der mit Placebo behandelten Patienten erhöhte TSH-Spiegel. Von den mit SEROQUEL 300 mg behandelten Patienten mit erhöhten TSH-Spiegeln hatten 3 gleichzeitig niedrige Werte von freiem T4 (freies T4
Bei etwa 0,7 % (26/3489) der SEROQUEL-Patienten kam es in Monotherapiestudien zu TSH-Anstiegen. Einige Patienten mit erhöhten TSH-Werten benötigten eine Schilddrüsenersatzbehandlung.
In allen Quetiapin-Studien betrug die Inzidenz von Veränderungen der Schilddrüsenhormone und des TSH1: Abnahme des freien T4 ( 5 mIU/l), 4,9 % (956/19412). Bei acht Patienten, bei denen TBG gemessen wurde, blieben die TBG-Spiegel unverändert.
Tabelle 8 zeigt die Inzidenz dieser Verschiebungen in placebokontrollierten klinischen Kurzzeitstudien.
In Placebo-kontrollierten Kurzzeit-Monotherapiestudien betrug die Inzidenz reziproker Verschiebungen von T3 und TSH 0,0 % für Quetiapin (1/4800) und Placebo (0/2190) und für T4 und TSH betrugen die Verschiebungen 0,1 % (7 /6154) für Quetiapin gegenüber 0,0 % (1/3007) für Placebo.
Kinder und Jugendliche
In akuten placebokontrollierten Studien bei Kindern und Jugendlichen mit Schizophrenie (Dauer 6 Wochen) oder bipolarer Manie (Dauer 3 Wochen) war die Inzidenz von Veränderungen der Schilddrüsenfunktionswerte zu jedem Zeitpunkt bei mit SEROQUEL 100 mg behandelten Patienten und bei mit Placebo behandelten Patienten bei erhöhtem TSH 2,9 % (8/280) vs. 0,7 % (1/138) und bei erniedrigtem Gesamtthyroxin 2,8 % (8/289) vs. 0 % (0/145). Von den mit SEROQUEL behandelten Patienten mit erhöhten TSH-Spiegeln hatte 1 gleichzeitig einen niedrigen freien T4-Spiegel am Ende der Behandlung.
Hyperprolaktinämie
Erwachsene
Während klinischer Studien mit Quetiapin kam es bei 3,6 % (158/4416) der mit Quetiapin behandelten Patienten im Vergleich zu 2,6 % (51/1968) unter Placebo zu Verschiebungen der Prolaktinspiegel auf einen klinisch signifikanten Wert.
Kinder und Jugendliche
In akuten placebokontrollierten Studien bei Kindern und Jugendlichen mit bipolarer Manie (Dauer 3 Wochen) oder Schizophrenie (Dauer 6 Wochen) war die Inzidenz von Verschiebungen der Prolaktinspiegel auf einen Wert (> 20 μg/L Männer; > 26 μg /L Frauen zu jedem Zeitpunkt) betrug 13,4 % (18/134) für SEROQUEL im Vergleich zu 4 % (3/75) für Placebo bei Männern und 8,7 % (9/104) für SEROQUEL 25 mg im Vergleich zu 0 % (0/39) für Placebo bei Frauen.
Wie andere Arzneimittel, die Dopamin-D2-Rezeptoren antagonisieren, erhöht SEROQUEL 300 mg bei einigen Patienten die Prolaktinspiegel, und die Erhöhung kann während einer chronischen Anwendung anhalten. Hyperprolaktinämie, unabhängig von der Ätiologie, kann hypothalamisches GnRH unterdrücken, was zu einer verringerten hypophysären Gonadotropinsekretion führt. Dies wiederum kann die Fortpflanzungsfunktion hemmen, indem es die gonadale Steroidogenese sowohl bei weiblichen als auch bei männlichen Patienten beeinträchtigt. Galaktorrhoe, Amenorrhoe, Gynäkomastie und Impotenz wurden bei Patienten berichtet, die Prolaktin-erhöhende Präparate erhielten. Langanhaltende Hyperprolaktinämie in Verbindung mit Hypogonadismus kann sowohl bei weiblichen als auch bei männlichen Probanden zu einer verminderten Knochendichte führen.
Gewebekulturexperimente weisen darauf hin, dass ungefähr ein Drittel der menschlichen Brustkrebserkrankungen in vitro Prolaktin-abhängig sind, ein Faktor von potenzieller Bedeutung, wenn die Verschreibung dieser Medikamente bei einer Patientin mit zuvor erkanntem Brustkrebs in Betracht gezogen wird. Wie bei Verbindungen, die die Prolaktinfreisetzung erhöhen, wurde in Kanzerogenitätsstudien an Mäusen und Ratten eine Neoplasie der Brustdrüse und der Inselzellen der Bauchspeicheldrüse (Mamma-Adenokarzinome, Hypophysen- und Pankreas-Adenome) beobachtet. Weder bisher durchgeführte klinische Studien noch epidemiologische Studien haben einen Zusammenhang zwischen der chronischen Verabreichung dieser Arzneimittelklasse und der Tumorentstehung beim Menschen gezeigt, aber die verfügbaren Beweise sind zu begrenzt, um schlüssig zu sein [siehe Nichtklinische Toxikologie ].
Potenzial für kognitive und motorische Beeinträchtigungen
Somnolenz war eine häufig berichtete Nebenwirkung bei Patienten, die mit SEROQUEL behandelt wurden, insbesondere während der 3- bis 5-tägigen Phase der anfänglichen Dosistitration. In Schizophrenie-Studien wurde Schläfrigkeit bei 18 % (89/510) der Patienten unter SEROQUEL 200 mg im Vergleich zu 11 % (22/206) der Placebo-Patienten berichtet. In Studien zur akuten bipolaren Manie mit SEROQUEL 200 mg als Monotherapie wurde bei 16 % (34/209) der Patienten unter SEROQUEL 300 mg Somnolenz berichtet, verglichen mit 4 % der Placebo-Patienten. In Studien zur akuten bipolaren Manie mit SEROQUEL als Zusatztherapie wurde Schläfrigkeit bei 34 % (66/196) der Patienten unter SEROQUEL im Vergleich zu 9 % (19/203) der Placebo-Patienten berichtet. In Studien zu bipolaren Depressionen wurde Schläfrigkeit bei 57 % (398/698) der Patienten unter SEROQUEL 50 mg im Vergleich zu 15 % (51/347) der Placebo-Patienten berichtet. Da SEROQUEL 25 mg das Urteilsvermögen, Denkvermögen oder motorische Fähigkeiten beeinträchtigen kann, sollten Patienten davor gewarnt werden, Aktivitäten durchzuführen, die geistige Wachsamkeit erfordern, wie z sie nicht negativ beeinflussen. Schläfrigkeit kann zu Stürzen führen.
Regulierung der Körpertemperatur
Obwohl dies nicht für SEROQUEL 100 mg berichtet wurde, wurde eine Störung der Fähigkeit des Körpers, die Körperkerntemperatur zu senken, auf Antipsychotika zurückgeführt. Bei der Verschreibung von SEROQUEL 300 mg für Patienten, die an Zuständen leiden, die zu einer Erhöhung der Körperkerntemperatur beitragen können, z. B. anstrengende körperliche Betätigung, extreme Hitzeeinwirkung, gleichzeitige Einnahme von Medikamenten mit anticholinerger Wirkung oder Dehydrierung, ist bei der Verschreibung von SEROQUEL 300 mg angemessene Vorsicht geboten.
Dysphagie
Dysmotilität und Aspiration des Ösophagus wurden mit dem Gebrauch von Antipsychotika in Verbindung gebracht. Aspirationspneumonie ist eine häufige Ursache für Morbidität und Mortalität bei älteren Patienten, insbesondere bei Patienten mit fortgeschrittener Alzheimer-Demenz. SEROQUEL 100 mg und andere Antipsychotika sollten bei Patienten mit einem Risiko für eine Aspirationspneumonie mit Vorsicht angewendet werden.
Abbruchsyndrom
Akute Entzugserscheinungen wie Schlaflosigkeit, Übelkeit und Erbrechen wurden nach abruptem Absetzen von atypischen Antipsychotika, einschließlich SEROQUEL, beschrieben. In placebokontrollierten klinischen Kurzzeitstudien zur Monotherapie mit SEROQUEL 25 mg XR, die eine Absetzphase umfassten, in der die Absetzsymptome bewertet wurden, betrug die aggregierte Inzidenz von Patienten, bei denen nach abruptem Absetzen ein oder mehrere Absetzsymptome auftraten, 12,1 % (241/1993) für SEROQUEL XR und 6,7 % (71/1065) für Placebo. Die Inzidenz der einzelnen Nebenwirkungen (dh Schlaflosigkeit, Übelkeit, Kopfschmerzen, Durchfall, Erbrechen, Schwindel und Reizbarkeit) überstieg in keiner Behandlungsgruppe 5,3 % und verschwand gewöhnlich 1 Woche nach Absetzen. Es wird ein schrittweiser Rückzug empfohlen. [sehen Verwendung in bestimmten Bevölkerungsgruppen ]
Anticholinerge (antimuskarinische) Wirkungen
Norquetiapin, ein aktiver Metabolit von Quetiapin, hat eine mäßige bis starke Affinität zu mehreren Muskarinrezeptor-Subtypen. Dies trägt zu anticholinergen Nebenwirkungen bei, wenn SEROQUEL 25 mg in therapeutischen Dosen, gleichzeitig mit anderen anticholinergen Arzneimitteln oder in Überdosierung eingenommen wird. SEROQUEL sollte bei Patienten, die Medikamente mit anticholinerger (antimuskarinischer) Wirkung erhalten, mit Vorsicht angewendet werden [siehe ÜBERDOSIS und KLINISCHE PHARMAKOLOGIE ].
Verstopfung war eine häufig berichtete Nebenwirkung bei mit Quetiapin behandelten Patienten und stellt einen Risikofaktor für Darmverschluss dar. Unter Quetiapin wurde über Darmverschluss berichtet, einschließlich tödlicher Fälle bei Patienten, die gleichzeitig mehrere Arzneimittel erhielten, die die Darmmotilität verringern.
SEROQUEL 300 mg sollte bei Patienten mit aktueller Diagnose oder Vorgeschichte von Harnverhalt, klinisch signifikanter Prostatahypertrophie, Verstopfung oder erhöhtem Augeninnendruck mit Vorsicht angewendet werden.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung (Medikamentenleitfaden) zu lesen.
Die Patienten sollten auf die folgenden Probleme hingewiesen und gebeten werden, ihren verschreibenden Arzt zu informieren, wenn diese während der Einnahme von SEROQUEL auftreten.
Erhöhte Sterblichkeit bei älteren Patienten mit Demenz-assoziierter Psychose Patienten und Betreuungspersonen sollten darauf hingewiesen werden, dass ältere Patienten mit Demenz-assoziierter Psychose, die mit atypischen Antipsychotika behandelt werden, im Vergleich zu Placebo einem erhöhten Sterberisiko ausgesetzt sind. Quetiapin ist für ältere Patienten mit demenzbedingter Psychose nicht zugelassen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Selbstmordgedanken und Verhaltensweisen
Patienten, ihre Familien und ihre Betreuer sollten ermutigt werden, auf das Auftreten von Angstzuständen, Unruhe, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Unruhe), Hypomanie, Manie und anderen ungewöhnlichen Verhaltensänderungen zu achten , Verschlechterung von Depressionen und Suizidgedanken, besonders früh während der Behandlung mit Antidepressiva und wenn die Dosis nach oben oder unten angepasst wird. Familien und Betreuer von Patienten sollten angewiesen werden, täglich auf das Auftreten solcher Symptome zu achten, da Änderungen abrupt sein können. Solche Symptome sollten dem verschreibenden Arzt oder medizinischen Fachpersonal des Patienten gemeldet werden, insbesondere wenn sie schwerwiegend sind, abrupt einsetzen oder nicht Teil der Symptome des Patienten waren. Symptome wie diese können mit einem erhöhten Risiko für Suizidgedanken und -verhalten verbunden sein und weisen auf die Notwendigkeit einer sehr engmaschigen Überwachung und möglicherweise einer Änderung der Medikation hin [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Malignes neuroleptisches Syndrom (NMS)
Patienten sollten angewiesen werden, ihrem Arzt alle Anzeichen oder Symptome zu melden, die mit NMS zusammenhängen könnten. Dazu können Muskelsteifheit und hohes Fieber gehören [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Hyperglykämie und Diabetes mellitus
Patienten sollten sich der Symptome von Hyperglykämie (hoher Blutzucker) und Diabetes mellitus bewusst sein. Patienten, bei denen Diabetes diagnostiziert wurde, die Risikofaktoren für Diabetes aufweisen oder die diese Symptome während der Behandlung entwickeln, sollten ihren Blutzucker zu Beginn und in regelmäßigen Abständen während der Behandlung kontrollieren lassen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Hyperlipidämie
Die Patienten sollten darauf hingewiesen werden, dass es zu einem Anstieg des Gesamtcholesterins, des LDL-Cholesterins und der Triglyceride und zu einem Abfall des HDL-Cholesterins kommen kann. Das Lipidprofil der Patienten sollte zu Beginn und in regelmäßigen Abständen während der Behandlung überwacht werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Gewichtszunahme
Die Patienten sollten darauf hingewiesen werden, dass es zu einer Gewichtszunahme kommen kann. Das Gewicht der Patienten sollte regelmäßig überwacht werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Orthostatische Hypotonie
Die Patienten sollten auf das Risiko einer orthostatischen Hypotonie hingewiesen werden (zu den Symptomen gehören Schwindel oder Benommenheit beim Aufstehen, was zu Stürzen führen kann), insbesondere während der anfänglichen Dosistitration und auch zu Zeiten der Wiederaufnahme der Behandlung oder Dosiserhöhungen [ sehen WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Erhöhter Blutdruck bei Kindern und Jugendlichen
Bei Kindern und Jugendlichen sollte der Blutdruck zu Beginn und in regelmäßigen Abständen während der Behandlung gemessen werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Leukopenie/Neutropenie
Patienten mit vorbestehenden niedrigen Leukozytenzahlen oder arzneimittelinduzierter Leukopenie/Neutropenie in der Vorgeschichte sollten darauf hingewiesen werden, dass sie während der Einnahme von SEROQUEL ihr Blutbild überwachen lassen sollten. Patienten sollten angewiesen werden, so schnell wie möglich mit ihrem Arzt zu sprechen, wenn sie Fieber, grippeähnliche Symptome, Halsschmerzen oder eine andere Infektion haben, da dies die Folge einer sehr niedrigen Leukozytenzahl sein könnte, die SEROQUEL 100 mg erfordern kann abgesetzt und/oder behandelt werden [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Beeinträchtigung der kognitiven und motorischen Leistung
Die Patienten sollten auf das Risiko von Somnolenz oder Sedierung (die zu Stürzen führen können) hingewiesen werden, insbesondere während der Phase der anfänglichen Dosistitration. Patienten sollten davor gewarnt werden, Aktivitäten durchzuführen, die geistige Wachsamkeit erfordern, wie z. B. das Führen eines Kraftfahrzeugs (einschließlich Autos) oder das Bedienen von Maschinen, bis sie hinreichend sicher sind, dass die Quetiapin-Therapie sie nicht nachteilig beeinflusst [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Hitzeeinwirkung und Austrocknung
Die Patienten sollten über angemessene Vorsichtsmaßnahmen zur Vermeidung von Überhitzung und Dehydration aufgeklärt werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Begleitmedikation
Wie bei anderen Medikamenten sollten Patienten angewiesen werden, ihren Arzt zu informieren, wenn sie verschreibungspflichtige oder rezeptfreie Medikamente einnehmen oder einnehmen möchten [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Schwangerschaft
Raten Sie schwangeren Frauen, ihren Arzt zu informieren, wenn sie schwanger werden oder beabsichtigen, während der Behandlung mit SEROQUEL schwanger zu werden. Weisen Sie die Patienten darauf hin, dass SEROQUEL bei einem Neugeborenen extrapyramidale Symptome und/oder Entzugssymptome (Agitiertheit, Hypertonie, Hypotonie, Tremor, Somnolenz, Atemnot und Fütterstörungen) verursachen kann. Weisen Sie die Patientinnen darauf hin, dass es ein Schwangerschaftsregister gibt, das den Ausgang der Schwangerschaft bei Frauen überwacht, die während der Schwangerschaft SEROQUEL 25 mg ausgesetzt waren [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Unfruchtbarkeit
Weisen Sie gebärfähige Frauen darauf hin, dass SEROQUEL die Fruchtbarkeit aufgrund eines Anstiegs der Prolaktinspiegel im Serum beeinträchtigen kann. Die Auswirkungen auf die Fertilität sind reversibel [vgl Verwendung in bestimmten Bevölkerungsgruppen ].
Notwendigkeit eines umfassenden Behandlungsprogramms
SEROQUEL ist als integraler Bestandteil eines Gesamtbehandlungsprogramms für Jugendliche mit Schizophrenie und pädiatrischer bipolarer Störung indiziert, das andere (psychologische, pädagogische und soziale) Maßnahmen umfassen kann. Wirksamkeit und Sicherheit von SEROQUEL 100 mg wurden bei pädiatrischen Patienten unter 13 Jahren bei Schizophrenie oder unter 10 Jahren bei bipolarer Manie nicht nachgewiesen. Eine angemessene schulische Unterbringung ist unerlässlich, und psychosoziale Interventionen sind oft hilfreich. Die Entscheidung, atypische Antipsychotika zu verschreiben, hängt von der Einschätzung des Arztes über die Chronizität und Schwere der Symptome des Patienten ab [siehe INDIKATIONEN ].
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Kanzerogenitätsstudien wurden an C57BL-Mäusen und Wistar-Ratten durchgeführt. Quetiapin wurde Mäusen in Dosen von 20, 75, 250 und 750 mg/kg über das Futter und Ratten in Dosen von 25, 75 und 250 mg/kg per Schlundsonde über zwei Jahre verabreicht. Diese Dosen entsprechen dem 0,1-, 0,5-, 1,5- und 4,5-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche (Mäuse) oder dem 0,3-, 1- und 3-fachen der MRHD, basierend auf mg/m2 Körperoberfläche (Ratten). Es gab statistisch signifikante Zunahmen follikulärer Adenome der Schilddrüse bei männlichen Mäusen bei Dosen, die das 1,5- und 4,5-fache der MRHD bezogen auf mg/m2 Körperoberfläche betrugen, und bei männlichen Ratten bei einer Dosis, die dem 3-fachen MRHD bezogen auf mg/m2 Körperoberfläche entsprach. Brustdrüsen-Adenokarzinome waren bei weiblichen Ratten bei allen getesteten Dosen statistisch signifikant erhöht (0,3-, 1- und 3-fache MRHD, basierend auf mg/m2 Körperoberfläche).
Adenome der follikulären Schilddrüsenzellen können aus einer chronischen Stimulation der Schilddrüse durch das Thyreoidea-stimulierende Hormon (TSH) resultieren, das aus einem verstärkten Metabolismus und der Clearance von Thyroxin durch die Leber von Nagetieren resultiert. Veränderungen von TSH, Thyroxin und Thyroxin-Clearance im Einklang mit diesem Mechanismus wurden in subchronischen Toxizitätsstudien an Ratten und Mäusen und in einer 1-Jahres-Toxizitätsstudie an Ratten beobachtet; Die Ergebnisse dieser Studien waren jedoch nicht endgültig. Die Relevanz des Anstiegs von Adenomen der Schilddrüsenfollikelzellen für das menschliche Risiko, durch welchen Mechanismus auch immer, ist unbekannt.
Es wurde gezeigt, dass Antipsychotika den Prolaktinspiegel bei Nagetieren chronisch erhöhen. Serummessungen in einer 1-Jahres-Toxizitätsstudie zeigten, dass Quetiapin die medianen Prolaktinspiegel im Serum bei männlichen bzw. weiblichen Ratten maximal um das 32- bzw. 13-Fache erhöhte. Bei Nagern wurde nach chronischer Verabreichung anderer Antipsychotika eine Zunahme von Mamma-Neoplasmen festgestellt, die als Prolaktin-vermittelt angesehen werden. Die Relevanz dieser erhöhten Inzidenz von Prolaktin-vermittelten Brustdrüsentumoren bei Ratten für das menschliche Risiko ist nicht bekannt [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Mutagenese
Quetiapin war in Standard-Genotoxizitätstests nicht mutagen oder klastogen. Das mutagene Potenzial von Quetiapin wurde im In-vitro-Ames-Bakterien-Genmutationstest und im In-vitro-Säugetier-Genmutationstest an Eierstockzellen des Chinesischen Hamsters getestet. Das klastogene Potenzial von Quetiapin wurde im In-vitro-Chromosomenaberrationsassay an kultivierten menschlichen Lymphozyten und im In-vivo-Knochenmark-Mikronukleus-Assay an Ratten bis zu 500 mg/kg getestet, was dem 6-fachen der empfohlenen Höchstdosis beim Menschen, basierend auf mg/m2, entspricht Körperoberfläche.
Beeinträchtigung der Fruchtbarkeit
Quetiapin verringerte die Paarung und Fertilität bei männlichen Sprague-Dawley-Ratten bei oralen Dosen von 50 und 150 mg/kg oder etwa dem 1- und 3-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche. Zu den medikamentenbedingten Wirkungen gehörten Verlängerungen des Paarungsintervalls und der Anzahl der Paarungen, die für eine erfolgreiche Befruchtung erforderlich sind. Diese Wirkungen wurden auch nach einem zweiwöchigen Zeitraum ohne Behandlung weiterhin beim 3-fachen der MRHD beobachtet. Die No-Effect-Dosis für beeinträchtigte Paarung und Fertilität bei männlichen Ratten betrug 25 mg/kg oder das 0,3-fache der MRHD-Dosis, basierend auf mg/m2 Körperoberfläche. Quetiapin beeinträchtigte die Paarung und Fertilität bei weiblichen Sprague-Dawley-Ratten bei einer oralen Dosis, die ungefähr dem 1-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche, entspricht. Zu den arzneimittelbedingten Wirkungen gehörten eine Abnahme der Paarungen und der Paarungen, die zu einer Schwangerschaft führten, sowie eine Verlängerung des Paarungsintervalls. Eine Zunahme unregelmäßiger Östruszyklen wurde bei Dosen von 10 und 50 mg/kg oder etwa dem 0,1- und 1-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche, beobachtet. Die Dosis ohne Wirkung bei weiblichen Ratten betrug 1 mg/kg oder das 0,01-fache der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschafts-Expositionsregister
Es gibt ein Schwangerschafts-Expositionsregister, das die Schwangerschaftsausgänge bei Frauen überwacht, die während der Schwangerschaft atypischen Antipsychotika, einschließlich SEROQUEL 50 mg, ausgesetzt waren. Gesundheitsdienstleister werden ermutigt, Patienten zu registrieren, indem sie sich unter 1-866-961-2388 oder online an das National Pregnancy Registry for Atypical Antipsychotics wenden http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/
Zusammenfassung der Risiken
Neugeborene, die während des dritten Trimesters Antipsychotika (einschließlich SEROQUEL) ausgesetzt waren, haben ein erhöhtes Risiko für extrapyramidale Symptome und/oder Entzugssymptome nach der Entbindung (siehe Klinische Überlegungen ). Insgesamt verfügbare Daten aus veröffentlichten epidemiologischen Studien an schwangeren Frauen, die Quetiapin ausgesetzt waren, haben kein arzneimittelbedingtes Risiko für schwerwiegende Geburtsfehler, Fehlgeburten oder unerwünschte Folgen für Mutter oder Fötus ergeben (siehe Daten ). Es bestehen Risiken für die Mutter im Zusammenhang mit einer unbehandelten Schizophrenie, Bipolar-I- oder schweren depressiven Störung und mit der Exposition gegenüber Antipsychotika, einschließlich SEROQUEL 200 mg, während der Schwangerschaft (siehe Klinische Überlegungen ).
In Tierstudien trat eine embryofetale Toxizität auf, einschließlich Verzögerungen der Skelettverknöcherung bei etwa dem 1- und 2-fachen der maximal empfohlenen Humandosis (MRHD) von 800 mg/Tag bei Ratten und Kaninchen und einer erhöhten Inzidenz von Handwurzel-/Tarsalflexur (geringfügige Weichteilanomalie) bei Kaninchenföten bei etwa dem 2-fachen der MRHD. Außerdem war das fetale Gewicht bei beiden Spezies verringert. Maternale Toxizität (beobachtet als verringertes Körpergewicht und/oder Tod) trat bei der 2-fachen MRHD bei Ratten und etwa der 1- bis 2-fachen MRHD bei Kaninchen auf.
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten für die angegebenen Populationen ist unbekannt. Alle Schwangerschaften haben ein Hintergrundrisiko für Geburtsfehler, Verlust oder andere nachteilige Folgen. In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwere Geburtsfehler und Fehlgeburten bei klinisch erkannten Schwangerschaften bei 2 bis 4 % bzw. 15 bis 20 %.
Klinische Überlegungen
Krankheitsbedingtes mütterliches und/oder fötales Risiko
Es besteht ein Risiko für die Mutter durch unbehandelte Schizophrenie oder Bipolar-I-Störung, einschließlich eines erhöhten Rückfallrisikos, Krankenhausaufenthalts und Suizids. Schizophrenie und Bipolar-I-Störung sind mit erhöhten unerwünschten perinatalen Folgen, einschließlich Frühgeburten, verbunden. Es ist nicht bekannt, ob dies eine direkte Folge der Krankheit oder anderer komorbider Faktoren ist.
Eine prospektive Längsschnittstudie folgte 201 schwangeren Frauen mit einer Vorgeschichte von schweren depressiven Störungen, die euthymisch waren und zu Beginn der Schwangerschaft Antidepressiva einnahmen. Frauen, die Antidepressiva während der Schwangerschaft absetzten, erlitten mit größerer Wahrscheinlichkeit einen Rückfall einer schweren Depression als Frauen, die Antidepressiva weiter einnahmen. Berücksichtigen Sie das Risiko einer unbehandelten Depression, wenn Sie die Behandlung mit Antidepressiva während der Schwangerschaft und nach der Geburt abbrechen oder ändern.
Fetale/neonatale Nebenwirkungen
Extrapyramidale und/oder Entzugserscheinungen, einschließlich Agitiertheit, Hypertonie, Hypotonie, Tremor, Somnolenz, Atemnot und Fütterstörungen, wurden bei Neugeborenen berichtet, die während des dritten Trimenons der Schwangerschaft Antipsychotika, einschließlich SEROQUEL 50 mg, ausgesetzt waren. Diese Symptome waren unterschiedlich stark ausgeprägt. Überwachen Sie Neugeborene auf extrapyramidale und/oder Entzugserscheinungen und behandeln Sie die Symptome entsprechend. Einige Neugeborene erholten sich ohne spezifische Behandlung innerhalb von Stunden oder Tagen; andere erforderten einen längeren Krankenhausaufenthalt.
Daten
Menschliche Daten
Veröffentlichte Daten aus Beobachtungsstudien, Geburtsregistern und Fallberichten über die Anwendung atypischer Antipsychotika während der Schwangerschaft zeigen keinen eindeutigen Zusammenhang mit Antipsychotika und schweren Geburtsfehlern. Eine retrospektive Kohortenstudie aus einer Medicaid-Datenbank mit 9258 Frauen, die während der Schwangerschaft Antipsychotika ausgesetzt waren, zeigte kein insgesamt erhöhtes Risiko für schwere Geburtsfehler.
Tierdaten
Wenn trächtige Ratten und Kaninchen während der Organogenese Quetiapin ausgesetzt wurden, gab es keine teratogene Wirkung auf Föten. Die Dosierungen betrugen 25, 50 und 200 mg/kg bei Ratten und 25, 50 und 100 mg/kg bei Kaninchen, was ungefähr dem 0,3-, 0,6- und 2-fachen (Ratten) und dem 0,6-, 1- und 2-fachen (Kaninchen) der MRHD entspricht Schizophrenie von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche. Es gab jedoch Hinweise auf eine embryofetale Toxizität, einschließlich Verzögerungen der Skelettossifikation bei etwa dem 1- und 2-fachen der MRHD von 800 mg/Tag sowohl bei Ratten als auch bei Kaninchen und einer erhöhten Inzidenz von Karpal-/Tarsalflexuren (geringfügige Weichteilanomalie) in Kaninchenföten bei etwa dem 2-fachen der MRHD. Außerdem war das fetale Gewicht bei beiden Spezies verringert. Maternale Toxizität (beobachtet als verringertes Körpergewicht und/oder Tod) trat bei der 2-fachen MRHD bei Ratten und etwa der 1-2-fachen MRHD (alle getesteten Dosen) bei Kaninchen auf.
In einer peri-/postnatalen Reproduktionsstudie an Ratten wurden keine arzneimittelbedingten Wirkungen beobachtet, wenn trächtige Muttertiere mit Quetiapin-Dosen behandelt wurden, die dem 0,01-, 0,1- und 0,2-Fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche, entsprachen. In einer vorläufigen peri-/postnatalen Studie kam es jedoch zu einem Anstieg des Todes von Föten und Jungtieren und zu einem Rückgang des mittleren Wurfgewichts beim 3-fachen der MRHD.
Stillzeit
Zusammenfassung der Risiken
Begrenzte Daten aus der veröffentlichten Literatur berichten über das Vorhandensein von Quetiapin in der menschlichen Muttermilch bei einer relativen Säuglingsdosis von
Weibchen und Männchen mit reproduktivem Potenzial
Unfruchtbarkeit
Frauen
Basierend auf der pharmakologischen Wirkung von Quetiapin (D2-Antagonismus) kann die Behandlung mit SEROQUEL zu einem Anstieg der Serum-Prolaktinspiegel führen, was zu einer reversiblen Verringerung der Fertilität bei gebärfähigen Frauen führen kann [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Pädiatrische Verwendung
Im Allgemeinen waren die Nebenwirkungen, die bei Kindern und Jugendlichen während der klinischen Studien beobachtet wurden, mit wenigen Ausnahmen denen bei Erwachsenen ähnlich. Anstiege des systolischen und diastolischen Blutdrucks traten bei Kindern und Jugendlichen auf und traten bei Erwachsenen nicht auf. Orthostatische Hypotonie trat häufiger bei Erwachsenen (4-7 %) im Vergleich zu Kindern und Jugendlichen ( WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Schizophrenie
Die Wirksamkeit und Sicherheit von SEROQUEL 50 mg bei der Behandlung von Schizophrenie bei Jugendlichen im Alter von 13 bis 17 Jahren wurden in einer 6-wöchigen, doppelblinden, placebokontrollierten Studie nachgewiesen [siehe INDIKATIONEN , DOSIERUNG UND ANWENDUNG , NEBENWIRKUNGEN , und Klinische Studien ].
Die Sicherheit und Wirksamkeit von SEROQUEL 300 mg bei pädiatrischen Patienten unter 13 Jahren mit Schizophrenie wurde nicht nachgewiesen.
Wartung
Die Sicherheit und Wirksamkeit von SEROQUEL in der Erhaltungstherapie der bipolaren Störung wurde bei pädiatrischen Patienten unter 18 Jahren nicht nachgewiesen. Die Sicherheit und Wirksamkeit von SEROQUEL in der Erhaltungstherapie der Schizophrenie wurde bei keiner Patientenpopulation, einschließlich pädiatrischer Patienten, nachgewiesen.
Bipolare Manie
Die Wirksamkeit und Sicherheit von SEROQUEL bei der Behandlung von Manie bei Kindern und Jugendlichen im Alter von 10 bis 17 Jahren mit Bipolar-I-Störung wurde in einer 3-wöchigen, doppelblinden, placebokontrollierten, multizentrischen Studie nachgewiesen [siehe INDIKATIONEN , DOSIERUNG UND ANWENDUNG , NEBENWIRKUNGEN , und Klinische Studien ]. Die Sicherheit und Wirksamkeit von SEROQUEL bei pädiatrischen Patienten unter 10 Jahren mit bipolarer Manie wurde nicht nachgewiesen.
Bipolare Depression
Sicherheit und Wirksamkeit von SEROQUEL 300 mg bei pädiatrischen Patienten unter 18 Jahren mit bipolarer Depression wurden nicht nachgewiesen. Eine klinische Studie mit SEROQUEL XR wurde bei Kindern und Jugendlichen (10-17 Jahre) mit bipolarer Depression durchgeführt, die Wirksamkeit wurde nicht nachgewiesen.
Einige Unterschiede in der Pharmakokinetik von Quetiapin wurden zwischen Kindern/Jugendlichen (10-17 Jahre) und Erwachsenen festgestellt. Gewichtsbereinigt waren die AUC und Cmax von Quetiapin bei Kindern und Jugendlichen um 41 % bzw. 39 % niedriger als bei Erwachsenen. Die Pharmakokinetik des aktiven Metaboliten Norquetiapin war nach Gewichtsanpassung bei Kindern/Jugendlichen und Erwachsenen ähnlich [siehe KLINISCHE PHARMAKOLOGIE ].
Geriatrische Verwendung
Von den ungefähr 3700 Patienten in klinischen Studien mit SEROQUEL 300 mg waren 7 % (232) 65 Jahre oder älter. Im Allgemeinen gab es keinen Hinweis auf eine unterschiedliche Verträglichkeit von SEROQUEL bei älteren Menschen im Vergleich zu jüngeren Erwachsenen. Dennoch sollte das Vorhandensein von Faktoren, die die pharmakokinetische Clearance verringern, die pharmakodynamische Reaktion auf SEROQUEL 200 mg verstärken oder eine schlechtere Verträglichkeit oder Orthostase verursachen könnten, dazu führen, dass eine niedrigere Anfangsdosis, eine langsamere Titration und eine sorgfältige Überwachung während der anfänglichen Dosierungsphase in Betracht gezogen werden Alten. Die mittlere Plasmaclearance von SEROQUEL 25 mg war bei älteren Patienten im Vergleich zu jüngeren Patienten um 30 % bis 50 % verringert [siehe KLINISCHE PHARMAKOLOGIE und DOSIERUNG UND ANWENDUNG ].
Nierenfunktionsstörung
Die klinische Erfahrung mit SEROQUEL bei Patienten mit eingeschränkter Nierenfunktion ist begrenzt [siehe KLINISCHE PHARMAKOLOGIE ].
Leberfunktionsstörung
Da Quetiapin weitgehend in der Leber metabolisiert wird, sind bei Patienten mit eingeschränkter Leberfunktion höhere Plasmaspiegel zu erwarten. Bei dieser Population wird eine niedrige Anfangsdosis von 25 mg/Tag empfohlen und die Dosis kann in Schritten von 25 mg/Tag bis 50 mg/Tag erhöht werden [siehe DOSIERUNG UND ANWENDUNG und KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Menschliche Erfahrung
In klinischen Studien wurde über das Überleben bei akuter Überdosierung von bis zu 30 Gramm Quetiapin berichtet. Bei den meisten Patienten mit Überdosierung traten keine Nebenwirkungen auf oder sie erholten sich vollständig von den berichteten Ereignissen. In einer klinischen Studie wurde über Todesfälle nach einer Überdosis von 13,6 Gramm Quetiapin allein berichtet. Im Allgemeinen handelte es sich bei den gemeldeten Anzeichen und Symptomen um solche, die aus einer Übertreibung der bekannten pharmakologischen Wirkungen des Arzneimittels resultierten, dh Schläfrigkeit, Sedierung, Tachykardie, Hypotonie und anticholinerge Toxizität, einschließlich Koma und Delirium. Bei Patienten mit vorbestehender schwerer Herz-Kreislauf-Erkrankung besteht möglicherweise ein erhöhtes Risiko für die Auswirkungen einer Überdosierung [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Ein Fall mit einer geschätzten Überdosierung von 9600 mg war mit Hypokaliämie und Herzblock ersten Grades verbunden. Nach Markteinführung wurden Fälle von QT-Verlängerung bei Überdosierung berichtet.
Management einer Überdosierung
Schaffen und erhalten Sie einen Atemweg und sorgen Sie für eine ausreichende Sauerstoffversorgung und Belüftung. Die kardiovaskuläre Überwachung sollte sofort beginnen und eine kontinuierliche elektrokardiographische Überwachung umfassen, um mögliche Arrhythmien zu erkennen.
Angemessene unterstützende Maßnahmen sind die tragende Säule des Managements. Wenden Sie sich für die aktuellsten Informationen zum Umgang mit einer Seroquel XR-Überdosierung an ein zertifiziertes regionales Giftinformationszentrum (1-800-222-1222).
KONTRAINDIKATIONEN
Überempfindlichkeit gegen Quetiapin oder einen der sonstigen Bestandteile der SEROQUEL XR-Formulierung. Anaphylaktische Reaktionen wurden bei mit SEROQUEL XR behandelten Patienten berichtet.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Der Wirkungsmechanismus von Quetiapin in den aufgeführten Indikationen ist unklar. Die Wirksamkeit von Quetiapin bei diesen Indikationen könnte jedoch durch eine Kombination aus Dopamin-Typ-2-(D2)- und Serotonin-Typ-2-(5HT2)-Antagonismus vermittelt werden. Der aktive Metabolit, N-Desalkylquetiapin (Norquetiapin), hat eine ähnliche Aktivität an D2, aber eine größere Aktivität an 5HT2A-Rezeptoren als die Ausgangssubstanz (Quetiapin).
Pharmakodynamik
Quetiapin und sein Metabolit Norquetiapin haben eine Affinität zu mehreren Neurotransmitterrezeptoren, wobei Norquetiapin mit einer höheren Affinität als Quetiapin im Allgemeinen bindet. Die Ki-Werte für Quetiapin und Norquetiapin bei Dopamin D1 sind 428/99,8 nM, bei D2 626/489 nM, bei Serotonin 5HT1A 1040/191 nM, bei 5HT2A 38/2,9 nM, bei Histamin H1 4,4/1,1 nM, bei Muscarin M1 1086/ 38,3 nM und bei adrenergen α1b 14,6/46,4 nM bzw. bei α2-Rezeptoren 617/1290 nM. Quetiapin und Norquetiapin haben keine nennenswerte Affinität zu den Benzodiazepin-Rezeptoren.
Auswirkung auf das QT-Intervall
In klinischen Studien war Quetiapin nicht mit einer anhaltenden Verlängerung der QT-Intervalle verbunden. Der QT-Effekt wurde jedoch nicht systematisch in einer gründlichen QT-Studie evaluiert. Nach der Markteinführung wurden Fälle von QT-Verlängerung bei Patienten berichtet, die Quetiapin überdosiert hatten [siehe ÜBERDOSIS ], bei Patienten mit Begleiterkrankungen und bei Patienten, die Arzneimittel einnehmen, von denen bekannt ist, dass sie ein Elektrolytungleichgewicht verursachen oder das QT-Intervall verlängern.
Pharmakokinetik
Erwachsene
Die Aktivität von Quetiapinfumarat ist hauptsächlich auf die Muttersubstanz zurückzuführen. Die Pharmakokinetik von Quetiapin nach Mehrfachgabe ist innerhalb des vorgeschlagenen klinischen Dosisbereichs dosisproportional, und eine Akkumulation von Quetiapin ist bei Mehrfachgabe vorhersehbar. Die Elimination von Quetiapin erfolgt hauptsächlich über die Leber mit einer mittleren terminalen Halbwertszeit von etwa 6 Stunden innerhalb des vorgeschlagenen klinischen Dosisbereichs. Es wird erwartet, dass Steady-State-Konzentrationen innerhalb von zwei Tagen nach der Verabreichung erreicht werden. Es ist unwahrscheinlich, dass Quetiapin den Metabolismus von Arzneimitteln beeinflusst, die durch Cytochrom-P450-Enzyme metabolisiert werden.
Kinder und Jugendliche
Im Steady State war die Pharmakokinetik der Muttersubstanz bei Kindern und Jugendlichen (im Alter von 10 bis 17 Jahren) ähnlich wie bei Erwachsenen. Bei Dosis- und Gewichtsanpassung waren die AUC und Cmax der Muttersubstanz jedoch bei Kindern und Jugendlichen um 41 % bzw. 39 % niedriger als bei Erwachsenen. Für den aktiven Metaboliten Norquetiapin waren AUC und Cmax bei Kindern und Jugendlichen um 45 % bzw. 31 % höher als bei Erwachsenen. Nach Anpassung an Dosis und Gewicht war die Pharmakokinetik des Metaboliten Norquetiapin bei Kindern und Jugendlichen und Erwachsenen ähnlich [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Absorption
Quetiapinfumarat wird nach oraler Verabreichung schnell resorbiert und erreicht maximale Plasmakonzentrationen innerhalb von 1,5 Stunden. Die Tablettenformulierung ist relativ zur Lösung zu 100 % bioverfügbar. Die Bioverfügbarkeit von Quetiapin wird durch die Einnahme zusammen mit Nahrung geringfügig beeinflusst, wobei die Cmax- und AUC-Werte um 25 % bzw. 15 % ansteigen.
Verteilung
Quetiapin wird mit einem scheinbaren Verteilungsvolumen von 10 ± 4 l/kg weit im Körper verteilt. In therapeutischen Konzentrationen ist es zu 83 % an Plasmaproteine gebunden. In vitro hatte Quetiapin keinen Einfluss auf die Bindung von Warfarin oder Diazepam an Humanserumalbumin. Weder Warfarin noch Diazepam wiederum veränderten die Bindung von Quetiapin.
Stoffwechsel und Ausscheidung
Nach einer oralen Einzeldosis von 14C-Quetiapin wurde weniger als 1 % der verabreichten Dosis als unveränderter Wirkstoff ausgeschieden, was darauf hindeutet, dass Quetiapin stark metabolisiert wird. Ungefähr 73 % bzw. 20 % der Dosis wurden im Urin bzw. im Stuhl wiedergefunden.
Quetiapin wird weitgehend von der Leber metabolisiert. Die wichtigsten Stoffwechselwege sind die Sulfoxidation zum Sulfoxid-Metaboliten und die Oxidation zum Ausgangssäure-Metaboliten; beide Metaboliten sind pharmakologisch inaktiv. In-vitro-Studien mit menschlichen Lebermikrosomen zeigten, dass das Isoenzym Cytochrom P450 3A4 am Metabolismus von Quetiapin zu seinem wichtigsten, aber inaktiven Sulfoxid-Metaboliten und am Metabolismus seines aktiven Metaboliten N-Desalkylquetiapin beteiligt ist.
Das Alter
Die orale Clearance von Quetiapin war bei älteren Patienten (≥ 65 Jahre, n = 9) im Vergleich zu jungen Patienten (n = 12) um 40 % verringert, und eine Dosisanpassung kann erforderlich sein [siehe DOSIERUNG UND ANWENDUNG ].
Geschlecht
Es gibt keinen geschlechtsspezifischen Einfluss auf die Pharmakokinetik von Quetiapin.
Wettrennen
Es gibt keinen Einfluss der Rasse auf die Pharmakokinetik von Quetiapin.
Rauchen
Rauchen hat keinen Einfluss auf die orale Clearance von Quetiapin.
Niereninsuffizienz
Patienten mit schwerer Nierenfunktionsstörung (Clcr = 10-30 ml/min/1,73 m2, n=8) hatten eine um 25 % niedrigere mittlere orale Clearance als gesunde Probanden (Clcr > 80 ml/min/1,73 m2, n=8), aber Die Quetiapin-Plasmakonzentrationen bei Patienten mit Niereninsuffizienz lagen innerhalb des Konzentrationsbereichs, der bei normalen Patienten beobachtet wurde, die die gleiche Dosis erhielten. Eine Dosisanpassung ist daher bei diesen Patienten nicht erforderlich [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Leberinsuffizienz
Patienten mit eingeschränkter Leberfunktion (n = 8) hatten eine um 30 % niedrigere mittlere orale Clearance von Quetiapin als gesunde Probanden. Bei zwei der 8 Patienten mit eingeschränkter Leberfunktion waren AUC und Cmax dreimal höher als die typischerweise bei gesunden Probanden beobachteten. Da Quetiapin extensiv von der Leber metabolisiert wird, werden höhere Plasmaspiegel bei Patienten mit eingeschränkter Leberfunktion erwartet, und eine Dosisanpassung kann erforderlich sein [siehe DOSIERUNG UND ANWENDUNG und VERWENDUNG IN BESTIMMTEN POPULATIONEN ].
Arzneimittelwechselwirkungsstudien
Die In-vivo-Bewertungen der Wirkung anderer Arzneimittel auf die Pharmakokinetik von Quetiapin sind in Tabelle 17 zusammengefasst [vgl DOSIERUNG UND ANWENDUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Daten zur In-vitro-Enzymhemmung deuten darauf hin, dass Quetiapin und 9 seiner Metaboliten eine geringe hemmende Wirkung auf den durch die Cytochrome CYP 1A2, 2C9, 2C19, 2D6 und 3A4 vermittelten In-vivo-Metabolismus haben würden. Quetiapin in Dosen von 750 mg/Tag hatte keinen Einfluss auf die Pharmakokinetik einer Einzeldosis von Antipyrin, Lithium oder Lorazepam (Tabelle 18) [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Tiertoxikologie und/oder Pharmakologie
Quetiapin verursachte eine dosisabhängige Zunahme der Pigmentablagerung in der Schilddrüse in Toxizitätsstudien an Ratten, die 4 Wochen oder länger dauerten, und in einer 2-jährigen Kanzerogenitätsstudie an Mäusen. In Rattenstudien betrugen die Dosen 10, 25, 50, 75, 150 und 250 mg/kg, was ungefähr dem 0,1-, 0,3-, 0,6-, 1-, 2- und 3-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche, entspricht , beziehungsweise. Die Dosen in der Karzinogenitätsstudie an Mäusen betrugen 20, 75, 250 und 750 mg/kg, was ungefähr dem 0,1-, 0,5-, 1,5- und 4,5-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körperoberfläche, entspricht. Es wurde gezeigt, dass die Pigmentablagerung bei Ratten irreversibel ist. Die Identität des Pigments konnte nicht bestimmt werden, es wurde jedoch festgestellt, dass es zusammen mit Quetiapin in follikulären Epithelzellen der Schilddrüse lokalisiert ist. Die funktionellen Auswirkungen und die Relevanz dieses Befunds für das menschliche Risiko sind nicht bekannt.
Bei Hunden, die Quetiapin für 6 oder 12 Monate, aber nicht für 1 Monat erhielten, traten bei einer Dosis von 100 mg/kg oder dem 4-fachen der MRHD von 800 fokale dreieckige Katarakte an der Verbindungsstelle der hinteren Nähte in der äußeren Kortikalis der Linse auf mg/Tag basierend auf mg/m2 Körperoberfläche. Dieser Befund kann auf die Hemmung der Cholesterinbiosynthese durch Quetiapin zurückzuführen sein. Quetiapin verursachte in Studien mit wiederholter Gabe an Hunden und Affen eine dosisabhängige Senkung des Plasmacholesterinspiegels; es gab jedoch keine Korrelation zwischen Plasmacholesterin und dem Vorhandensein von Katarakten bei einzelnen Hunden. Das Auftreten von delta-8-Cholestanol im Plasma steht im Einklang mit der Hemmung eines späten Stadiums der Cholesterinbiosynthese bei diesen Spezies. In einer speziellen Studie an mit Quetiapin behandelten weiblichen Hunden wurde auch eine 25%ige Verringerung des Cholesteringehalts der äußeren Kortikalis der Linse beobachtet. Arzneimittelbedingte Katarakte wurden bei keiner anderen Spezies beobachtet; In einer 1-Jahres-Studie an Affen wurde jedoch bei 2/7 Frauen bei einer Dosis von 225 mg/kg oder dem 5,5-fachen der MRHD von 800 mg/Tag, basierend auf mg/m2 Körper, ein gestreiftes Aussehen der vorderen Linsenoberfläche festgestellt Oberfläche.
Klinische Studien
Schizophrenie
Kurzzeitversuche-Erwachsene
Die Wirksamkeit von SEROQUEL 50 mg bei der Behandlung von Schizophrenie wurde in 3 kontrollierten Kurzzeitstudien (6 Wochen) mit stationären Patienten mit Schizophrenie nachgewiesen, die die DSM III-R-Kriterien für Schizophrenie erfüllten. Obwohl in einer der drei Studien ein Arm mit einer einzelnen Haloperidol-Festdosis als Vergleichsbehandlung eingeschlossen wurde, war diese Gruppe mit einer einzelnen Haloperidol-Dosis nicht geeignet, um einen zuverlässigen und gültigen Vergleich zwischen SEROQUEL 200 mg und Haloperidol zu ermöglichen.
In diesen Studien wurden mehrere Instrumente zur Bewertung psychiatrischer Anzeichen und Symptome verwendet, darunter die Brief Psychiatric Rating Scale (BPRS), eine aus mehreren Elementen bestehende Bestandsaufnahme der allgemeinen Psychopathologie, die traditionell zur Bewertung der Auswirkungen einer medikamentösen Behandlung bei Schizophrenie verwendet wird. Das BPRS-Psychose-Cluster (konzeptionelle Desorganisation, halluzinatorisches Verhalten, Misstrauen und ungewöhnliche Gedankeninhalte) gilt als besonders nützliche Untergruppe zur Beurteilung aktiv psychotischer schizophrener Patienten. Eine zweite traditionelle Bewertung, der Clinical Global Impression (CGI), spiegelt den Eindruck eines erfahrenen Beobachters, der mit den Manifestationen der Schizophrenie vollständig vertraut ist, über den klinischen Gesamtzustand des Patienten wider.
Die Ergebnisse der Versuche folgen:
Die primären Wirksamkeitsergebnisse dieser drei Studien zur Behandlung von Schizophrenie bei Erwachsenen sind in Tabelle 19 dargestellt.
Die Untersuchung von Bevölkerungsuntergruppen (Rasse, Geschlecht und Alter) ergab keine unterschiedliche Reaktionsfähigkeit auf der Grundlage von Rasse oder Geschlecht, mit einer offensichtlich größeren Wirkung bei Patienten unter 40 Jahren im Vergleich zu Patienten über 40. Die klinische Bedeutung von dieser Befund ist unbekannt.
Jugendliche (13-17 Jahre)
Die Wirksamkeit von SEROQUEL 300 mg bei der Behandlung von Schizophrenie bei Jugendlichen (im Alter von 13 bis 17 Jahren) wurde in einer 6-wöchigen, doppelblinden, placebokontrollierten Studie (Studie 4) nachgewiesen. Patienten, die die DSM-IV-Diagnosekriterien für Schizophrenie erfüllten, wurden randomisiert einer von drei Behandlungsgruppen zugeteilt: SEROQUEL 400 mg/Tag (n=73), SEROQUEL 800 mg/Tag (n=74) oder Placebo (n=75). Die Studienmedikation wurde mit 50 mg/Tag begonnen und am Tag 2 auf 100 mg/Tag erhöht (aufgeteilt und zwei- oder dreimal täglich verabreicht). Anschließend wurde die Dosis auf die Zieldosis von 400 mg/Tag oder 800 mg/Tag in Schritten von 100 mg/Tag titriert, aufgeteilt und zwei- oder dreimal täglich verabreicht. Die primäre Wirksamkeitsvariable war die mittlere Veränderung gegenüber dem Ausgangswert auf der gesamten positiven und negativen Syndromskala (PANSS).
SEROQUEL mit 400 mg/Tag und 800 mg/Tag war Placebo bei der Reduktion des PANSS-Gesamtscores überlegen. Die primären Wirksamkeitsergebnisse dieser Studie bei der Behandlung von Schizophrenie bei Jugendlichen sind in Tabelle 19 dargestellt.
Bipolare Störung
Bipolare I-Störung, manische oder gemischte Episoden
Erwachsene
Die Wirksamkeit von SEROQUEL bei der akuten Behandlung von manischen Episoden wurde in 3 placebokontrollierten Studien bei Patienten nachgewiesen, die die DSM-IV-Kriterien für eine Bipolar-I-Störung mit manischen Episoden erfüllten. Diese Studien schlossen Patienten mit oder ohne psychotische Merkmale ein und schlossen Patienten mit Rapid Cycling und gemischten Episoden aus. Von diesen Studien waren 2 eine Monotherapie (12 Wochen) und 1 eine Zusatztherapie (3 Wochen) zu entweder Lithium oder Divalproex. Die wichtigsten Ergebnisse dieser Studien waren die Veränderung des Young Mania Rating Scale (YMRS)-Scores gegenüber dem Ausgangswert nach 3 und 12 Wochen für die Monotherapie und nach 3 Wochen für die Zusatztherapie. Eine Zusatztherapie ist definiert als die gleichzeitige Einleitung oder anschließende Verabreichung von SEROQUEL 100 mg mit Lithium oder Divalproex.
Das primäre Bewertungsinstrument, das in diesen Studien zur Bewertung manischer Symptome verwendet wurde, war YMRS, eine 11-Punkte-Skala, die von Klinikern bewertet wurde und traditionell zur Bewertung des Grades manischer Symptomatik verwendet wird (Reizbarkeit, störendes/aggressives Verhalten, Schlaf, gehobene Stimmung, Sprache, erhöhte Aktivität, sexuelles Interesse, Sprach-/Denkstörung, Gedankeninhalt, Aussehen und Einsicht) in einem Bereich von 0 (keine manischen Züge) bis 60 (maximale Punktzahl).
Die Ergebnisse der Versuche folgen:
Monotherapie
Die Wirksamkeit von SEROQUEL bei der Akutbehandlung der bipolaren Manie wurde in 2 placebokontrollierten Studien nachgewiesen. In zwei 12-wöchigen Studien (n = 300, n = 299), in denen SEROQUEL mit Placebo verglichen wurde, war SEROQUEL 300 mg Placebo bei der Reduzierung des YMRS-Gesamtscores in Woche 3 und 12 überlegen. Die Mehrheit der Patienten in diesen Studien nahm SEROQUEL 100 mg ein wurden in einem Bereich zwischen 400 mg/Tag und 800 mg pro Tag dosiert (Studien 1 und 2 in Tabelle 20).
Zusatztherapie
In dieser 3-wöchigen placebokontrollierten Studie wurden 170 Patienten mit bipolarer Manie (YMRS ≥20) randomisiert und erhielten SEROQUEL 100 mg oder Placebo als Zusatzbehandlung zu Lithium oder Divalproex. Patienten können vor der Randomisierung eine angemessene Behandlung mit Lithium oder Divalproex erhalten haben oder auch nicht. SEROQUEL 50 mg war Placebo überlegen, wenn es zu Lithium oder Divalproex allein bei der Reduzierung des YMRS-Gesamtscores hinzugefügt wurde (Studie 3 in Tabelle 20).
Die Mehrheit der Patienten in dieser Studie, die SEROQUEL 25 mg einnahmen, erhielt eine Dosis zwischen 400 mg/Tag und 800 mg pro Tag. In einer ähnlich angelegten Studie (n=200) war SEROQUEL mit einer Verbesserung der YMRS-Scores verbunden, zeigte jedoch keine Überlegenheit gegenüber Placebo, möglicherweise aufgrund eines höheren Placebo-Effekts.
Die primären Wirksamkeitsergebnisse dieser Studien zur Behandlung von Manie bei Erwachsenen sind in Tabelle 20 dargestellt.
Kinder und Jugendliche (10-17 Jahre)
Die Wirksamkeit von SEROQUEL bei der akuten Behandlung von manischen Episoden im Zusammenhang mit einer Bipolar-I-Störung bei Kindern und Jugendlichen (im Alter von 10 bis 17 Jahren) wurde in einer 3-wöchigen, doppelblinden, placebokontrollierten, multizentrischen Studie nachgewiesen (Studie 4 in Tabelle 20). Patienten, die die DSM-IV-Diagnosekriterien für eine manische Episode erfüllten, wurden in eine von drei Behandlungsgruppen randomisiert: SEROQUEL 400 mg/Tag (n=95), SEROQUEL 600 mg/Tag (n=98) oder Placebo (n=91) . Die Studienmedikation wurde mit 50 mg/Tag begonnen und am Tag 2 auf 100 mg/Tag erhöht (geteilte Dosen zwei- oder dreimal täglich). Anschließend wurde die Dosis auf eine Zieldosis von 400 mg/Tag oder 600 mg/Tag unter Verwendung von Schritten von 100 mg/Tag titriert, die in geteilten Dosen zwei- oder dreimal täglich verabreicht wurden. Die primäre Wirksamkeitsvariable war die mittlere Veränderung des YMRS-Gesamtscores gegenüber dem Ausgangswert.
SEROQUEL 400 mg/Tag und 600 mg/Tag waren Placebo bei der Verringerung des YMRS-Gesamtscores überlegen (Tabelle 20).
Bipolare Störung, depressive Episoden
Erwachsene
Die Wirksamkeit von SEROQUEL 25 mg zur akuten Behandlung depressiver Episoden im Zusammenhang mit einer bipolaren Störung wurde in 2 identisch konzipierten 8-wöchigen, randomisierten, doppelblinden, placebokontrollierten Studien (N = 1045) nachgewiesen (Studien 5 und 6 in Tabelle 21). . Diese Studien umfassten Patienten mit Bipolar-I- oder -II-Störung und solche mit oder ohne Rapid-Cycling-Kurs. Patienten, die auf SEROQUEL 200 mg randomisiert wurden, erhielten feste Dosen von entweder 300 mg oder 600 mg einmal täglich.
Das primäre Bewertungsinstrument zur Bewertung depressiver Symptome in diesen Studien war die Montgomery-Asberg-Depressionsbewertungsskala (MADRS), eine 10-Punkte-Skala, die von Ärzten mit Werten von 0 bis 60 bewertet wurde. Der primäre Endpunkt in beiden Studien war die Veränderung von Ausgangswert des MADRS-Scores in Woche 8. In beiden Studien war SEROQUEL 50 mg Placebo bei der Senkung des MADRS-Scores überlegen. Eine Verbesserung der Symptome, gemessen an der Veränderung des MADRS-Scores im Vergleich zu Placebo, wurde in beiden Studien an Tag 8 (Woche 1) und danach beobachtet. In diesen Studien wurde bei der 600-mg-Dosis kein zusätzlicher Nutzen festgestellt. Bei der 300-mg-Dosisgruppe wurden statistisch signifikante Verbesserungen gegenüber Placebo bei der allgemeinen Lebensqualität und der Zufriedenheit in Bezug auf verschiedene Funktionsbereiche beobachtet, gemessen mit dem Q-LES-Q(SF).
Die primären Wirksamkeitsergebnisse dieser Studien bei der akuten Behandlung depressiver Episoden im Zusammenhang mit einer bipolaren Störung bei Erwachsenen sind in Tabelle 21 dargestellt.
Erhaltungsbehandlung als Ergänzung zu Lithium oder Divalproex
Die Wirksamkeit von SEROQUEL 100 mg bei der Erhaltungstherapie der Bipolar-I-Störung wurde in 2 placebokontrollierten Studien bei Patienten (n=1326) nachgewiesen, die die DSM-IV-Kriterien für Bipolar-I-Störung erfüllten (Studien 7 und 8 in Abbildungen 1 und 2). Die Studien umfassten Patienten, deren jüngste Episode manisch, depressiv oder gemischt war, mit oder ohne psychotische Merkmale. In der Open-Label-Phase mussten die Patienten mindestens 12 Wochen lang stabil auf SEROQUEL plus Lithium oder Divalproex sein, um randomisiert zu werden. Im Durchschnitt wurden die Patienten für 15 Wochen stabilisiert. In der Randomisierungsphase setzten die Patienten die Behandlung mit Lithium oder Divalproex fort und erhielten randomisiert entweder SEROQUEL (verabreicht zweimal täglich mit insgesamt 400 mg/Tag bis 800 mg/Tag) oder Placebo. Etwa 50 % der Patienten hatten die SEROQUEL-Gruppe bis Tag 280 und 50 % der Placebo-Gruppe bis Tag 117 der doppelblinden Behandlung abgebrochen. Der primäre Endpunkt in diesen Studien war die Zeit bis zum Wiederauftreten eines Stimmungsereignisses (manische, gemischte oder depressive Episode). Ein Stimmungsereignis wurde als Einleitung einer Medikation oder Krankenhausaufenthalt wegen einer Stimmungsepisode definiert; YMRS-Score≥20 oder MADRS-Score≥20 bei 2 aufeinanderfolgenden Bewertungen; oder Studienabbruch aufgrund eines Stimmungsereignisses (Abbildung 1 und Abbildung 2).
In beiden Studien war SEROQUEL dem Placebo bei der Verlängerung der Zeit bis zum Wiederauftreten jeglicher Stimmungsschwankungen überlegen. Der Behandlungseffekt war für die Verlängerung der Zeit bis zum Wiederauftreten sowohl manischer als auch depressiver Episoden vorhanden. Die Wirkung von SEROQUEL war unabhängig von einer bestimmten Untergruppe (zugewiesener Stimmungsstabilisator, Geschlecht, Alter, Rasse, jüngste bipolare Episode oder Rapid-Cycling-Kurs).
Abbildung 1: Kaplan-Meier-Kurven der Zeit bis zum Wiederauftreten eines Stimmungsereignisses (Studie 7)
Abbildung 2: Kaplan-Meier-Kurven der Zeit bis zum Wiederauftreten eines Stimmungsereignisses (Studie 8)
INFORMATIONEN ZUM PATIENTEN
SEROQUEL (SER-oh-kwell)(Quetiapinfumarat) Tabletten
Lesen Sie diesen Arzneimittelleitfaden, bevor Sie mit der Einnahme von SEROQUEL beginnen und jedes Mal, wenn Sie eine Nachfüllung erhalten. Möglicherweise gibt es neue Informationen. Diese Informationen ersetzen nicht das Gespräch mit Ihrem Gesundheitsdienstleister über Ihren Gesundheitszustand oder Ihre Behandlung.
Was sind die wichtigsten Informationen, die ich über SEROQUEL wissen sollte?
SEROQUEL 100 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Rufen Sie sofort einen Arzt an, wenn Sie oder Ihr Familienmitglied eines der folgenden Symptome haben, insbesondere wenn sie neu oder schlimmer sind oder Sie beunruhigen:
Was muss ich sonst noch über Antidepressiva wissen?
Was ist SEROQUEL 300mg?
SEROQUEL 100 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung von:
Es ist nicht bekannt, ob SEROQUEL 100 mg bei Kindern unter 10 Jahren sicher und wirksam ist.
Was sollte ich meinem Arzt vor der Einnahme von SEROQUEL 100 mg mitteilen?
Bevor Sie SEROQUEL 200 mg einnehmen, informieren Sie Ihren Arzt, wenn Sie Folgendes haben oder hatten:
Informieren Sie den Arzt über alle Arzneimittel, die Sie einnehmen oder kürzlich eingenommen haben einschließlich verschreibungspflichtiger Arzneimittel, rezeptfreier Arzneimittel, Kräuterergänzungen und Vitamine.
SEROQUEL 300 mg und andere Arzneimittel können sich gegenseitig beeinflussen und schwerwiegende Nebenwirkungen verursachen. SEROQUEL 200 mg kann die Wirkungsweise anderer Arzneimittel beeinflussen, und andere Arzneimittel können die Wirkungsweise von SEROQUEL 200 mg beeinflussen.
Teilen Sie Ihrem medizinischen Betreuer mit, wenn Sie einen Drogenscreening im Urin haben, da SEROQUEL Ihre Testergebnisse beeinflussen kann. Teilen Sie den Testpersonen mit, dass Sie SEROQUEL einnehmen.
Wie sollte ich SEROQUEL einnehmen?
Was sollte ich während der Einnahme von SEROQUEL 100 mg vermeiden?
Welche Nebenwirkungen kann SEROQUEL 300 mg haben?
SEROQUEL 200 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Bei manchen Personen, die SEROQUEL einnehmen, kann es zu Blutzuckererhöhungen kommen. Extrem hoher Blutzucker kann zu Koma oder Tod führen. Wenn Sie an Diabetes oder Risikofaktoren für Diabetes leiden (z. B. Übergewicht oder Diabetes in der Familie), sollte Ihr Arzt Ihren Blutzuckerspiegel vor Beginn der Behandlung mit SEROQUEL 50 mg und während der Behandlung kontrollieren. Rufen Sie Ihren Arzt an, wenn Sie während der Einnahme von SEROQUEL eines dieser Symptome eines hohen Blutzuckers (Hyperglykämie) haben:
Zu den häufigsten Nebenwirkungen von SEROQUEL 50 mg gehören:
Bei Erwachsenen:
Bei Kindern und Jugendlichen:
Dies sind nicht alle möglichen Nebenwirkungen von SEROQUEL. Weitere Informationen erhalten Sie von Ihrem Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie sollte ich SEROQUEL 100 mg aufbewahren?
Allgemeine Informationen zur sicheren und wirksamen Anwendung von SEROQUEL
Medikamente werden manchmal für andere als die in einem Medikationsleitfaden aufgeführten Zwecke verschrieben. Verwenden Sie SEROQUEL nicht für einen Zustand, für den es nicht verschrieben wurde. Geben Sie SEROQUEL nicht an andere Personen weiter, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Dieser Arzneimittelleitfaden fasst die wichtigsten Informationen zu SEROQUEL zusammen. Wenn Sie weitere Informationen wünschen, wenden Sie sich an Ihren Arzt. Sie können Ihren Apotheker oder Gesundheitsdienstleister nach Informationen über SEROQUEL 100 mg fragen, die für Angehörige der Gesundheitsberufe bestimmt sind.
Weitere Informationen finden Sie unter www.SEROQUEL.com , oder rufen Sie 1-800-236-9933 an.
Welche Inhaltsstoffe enthält SEROQUEL 200 mg?
Wirkstoff: Quetiapinfumarat
Inaktive Zutaten: Povidon, dibasisches Dicalciumphosphatdihydrat, mikrokristalline Cellulose, Natriumstärkeglycolat, Lactosemonohydrat, Magnesiumstearat, Hypromellose, Polyethylenglycol und Titandioxid. Die 25-mg-Tabletten enthalten rotes und gelbes Eisenoxid. Die 100-mg- und 400-mg-Tabletten enthalten nur gelbes Eisenoxid.
Dieser Medikationsleitfaden wurde von der US Food and Drug Administration genehmigt.
Was ist Sinequan und wie wird es angewendet?
Sinequan ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Depressionen und Angstzuständen. Sinequan kann allein oder zusammen mit anderen Medikamenten verwendet werden.
Sinequan 75 mg gehört zu einer Klasse von Arzneimitteln, die als Antidepressiva, TCAs, bezeichnet werden.
Es ist nicht bekannt, ob Sinequan bei Kindern unter 12 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Sinequan 25 mg haben?
Sinequan 25 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Sinequan gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Sinequan. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Suizidalität und Antidepressiva
Antidepressiva erhöhten im Vergleich zu Placebo das Risiko für suizidales Denken und Verhalten (Suizidalität) bei Kindern, Jugendlichen und jungen Erwachsenen in Kurzzeitstudien zu Major Depression (MDD) und anderen psychiatrischen Erkrankungen. Jeder, der die Anwendung von Sinequan oder anderen Antidepressiva bei einem Kind, Jugendlichen oder jungen Erwachsenen in Betracht zieht, muss dieses Risiko mit der klinischen Notwendigkeit abwägen. Kurzzeitstudien zeigten bei Erwachsenen über 24 Jahren keine Erhöhung des Suizidalitätsrisikos mit Antidepressiva im Vergleich zu Placebo; Bei Erwachsenen ab 65 Jahren war das Risiko unter Antidepressiva im Vergleich zu Placebo geringer. Depressionen und bestimmte andere psychiatrische Störungen sind selbst mit einem erhöhten Suizidrisiko verbunden. Patienten jeden Alters, die mit einer antidepressiven Therapie begonnen werden, sollten angemessen überwacht und engmaschig auf klinische Verschlechterung, Suizidalität oder ungewöhnliche Verhaltensänderungen beobachtet werden. Familien und Betreuer sollten auf die Notwendigkeit einer genauen Beobachtung und Kommunikation mit dem verschreibenden Arzt hingewiesen werden. Sinequan ist nicht für die Anwendung bei pädiatrischen Patienten zugelassen. (Sehen WARNUNGEN : Klinische Verschlechterung und Suizidrisiko , INFORMATIONEN ZUM PATIENTEN , und VORSICHTSMASSNAHMEN : Pädiatrische Verwendung )
BEZEICHNUNG
SINEQUAN® (Doxepin-Hydrochlorid) gehört zu einer Klasse von Psychotherapeutika, die als trizyklische Dibenzoxepin-Verbindungen bekannt sind. Die Molekularformel der Verbindung ist C19H21NO·HCl mit einem Molekulargewicht von 316. Sie ist ein weißer kristalliner Feststoff, der in Wasser, niederen Alkoholen und Chloroform leicht löslich ist.
Inerte Inhaltsstoffe für die Kapselformulierungen sind: Hartgelatinekapseln (die Blue 1, Red 3, Red 40, Yellow 10 und andere inerte Inhaltsstoffe enthalten können); Magnesiumstearat; Natriumlaurylsulfat; Stärke.
Inerte Inhaltsstoffe für die orale Konzentratformulierung sind: Glycerin; Methylparaben; Pfefferminz Öl; Propylparaben; Wasser.
Chemie
SINEQUAN (Doxepin HCl) ist ein Dibenzoxepin-Derivat und das erste einer Familie trizyklischer Psychotherapeutika. Insbesondere ist es ein Isomerengemisch aus: 1-Propanamin, 3-Dibenz[b,e]oxepin-11(6H)yliden-N,N-dimethyl-, Hydrochlorid.
INDIKATIONEN
SINEQUAN wird empfohlen zur Behandlung von:
Zu den Zielsymptomen der Psychoneurose, die besonders gut auf SINEQUAN 10mg ansprechen, gehören Angst, Anspannung, Depression, somatische Symptome und Sorgen, Schlafstörungen, Schuldgefühle, Energielosigkeit, Angst, Besorgnis und Sorge.
Klinische Erfahrungen haben gezeigt, dass SINEQUAN 25 mg auch bei älteren Patienten sicher und gut verträglich ist. Aufgrund fehlender klinischer Erfahrung bei Kindern und Jugendlichen wird SINEQUAN nicht für die Anwendung bei Kindern unter 12 Jahren empfohlen.
DOSIERUNG UND ANWENDUNG
Für die meisten Patienten mit leichter bis mittelschwerer Erkrankung wird eine anfängliche Tagesdosis von 75 mg empfohlen. Die Dosierung kann anschließend in angemessenen Abständen und je nach individueller Reaktion erhöht oder verringert werden. Der übliche optimale Dosisbereich beträgt 75 mg/Tag bis 150 mg/Tag.
Bei schwerer erkrankten Patienten können höhere Dosen erforderlich sein, mit anschließender schrittweiser Erhöhung auf 300 mg/Tag, falls erforderlich. Ein zusätzlicher therapeutischer Effekt wird selten erreicht, wenn eine Dosis von 300 mg/Tag überschritten wird.
Bei Patienten mit sehr leichter Symptomatologie oder emotionalen Symptomen, die eine organische Erkrankung begleiten, können niedrigere Dosen ausreichen. Einige dieser Patienten wurden mit Dosen von nur 25–50 mg/Tag kontrolliert.
Die gesamte Tagesdosis von SINEQUAN 75 mg kann nach einem geteilten oder einmal täglichen Dosierungsschema verabreicht werden. Bei einmal täglicher Gabe beträgt die empfohlene Höchstdosis 150 mg/Tag. Diese Dosis kann vor dem Schlafengehen verabreicht werden. Die Kapselstärke von 150 mg ist nur für die Erhaltungstherapie vorgesehen und wird nicht für den Behandlungsbeginn empfohlen.
Die Anti-Angst-Wirkung tritt vor der antidepressiven Wirkung auf. Eine optimale antidepressive Wirkung ist möglicherweise erst nach zwei bis drei Wochen erkennbar.
WIE GELIEFERT
SINEQUAN ist als Kapseln erhältlich, die Doxepin-HCl enthalten, das entspricht:
10mg – 100er ( NDC 0049-5340-66) 25mg – 100er ( NDC 05350-66) 50mg – 100er ( NDC 05360-66) 75mg – 100er ( NDC 05390-66) 100mg – 100er ( NDC 05380-66) 150mg – 50er ( NDC 0049-5370-50)
SINEQUAN Oralkonzentrat ist in 120-ml-Flaschen erhältlich ( NDC 0049-5100-47) mit einer dazugehörigen Pipette, die auf 5 mg, 10 mg, 15 mg, 20 mg und 25 mg kalibriert ist. Jeder ml enthält Doxepin-HCl entsprechend 10 mg Doxepin. Unmittelbar vor der Anwendung sollte SINEQUAN 25 mg Konzentrat zum Einnehmen mit etwa 120 ml Wasser, Voll- oder Magermilch oder Orangen-, Grapefruit-, Tomaten-, Pflaumen- oder Ananassaft verdünnt werden. SINEQUAN 25 mg orales Konzentrat ist mit einer Reihe von kohlensäurehaltigen Getränken physikalisch nicht kompatibel. Für Patienten, die eine antidepressive Therapie benötigen und Methadon erhalten, können SINEQUAN 10 mg orales Konzentrat und Methadonsirup mit Gatorade®, Limonade, Orangensaft, Zuckerwasser, Tang® oder Wasser gemischt werden; aber nicht mit Traubensaft. Die Herstellung und Lagerung von Massenverdünnungen wird nicht empfohlen.
Vertrieb durch: Roerig, Division of Pfizer Inc, NY, NY 10017. Juni 2014
NEBENWIRKUNGEN
HINWEIS: Einige der unten aufgeführten Nebenwirkungen wurden bei der Anwendung von SINEQUAN 10 mg nicht speziell berichtet. Aufgrund der engen pharmakologischen Ähnlichkeiten zwischen den Trizyklika sollten die Reaktionen jedoch bei der Verschreibung von SINEQUAN (Doxepin-HCl) berücksichtigt werden.
Anticholinerge Wirkungen
Mundtrockenheit, verschwommenes Sehen, Verstopfung und Harnverhalt wurden berichtet. Wenn sie bei fortgesetzter Therapie nicht abklingen oder schwerwiegend werden, kann es erforderlich sein, die Dosierung zu reduzieren.
Auswirkungen auf das zentrale Nervensystem
Schläfrigkeit ist die am häufigsten beobachtete Nebenwirkung. Dies verschwindet tendenziell, wenn die Therapie fortgesetzt wird. Andere selten berichtete ZNS-Nebenwirkungen sind Verwirrtheit, Orientierungslosigkeit, Halluzinationen, Taubheitsgefühl, Parästhesien, Ataxie, extrapyramidale Symptome, Krampfanfälle, tardive Dyskinesie und Tremor.
Herz-Kreislauf
Gelegentlich wurde über kardiovaskuläre Wirkungen einschließlich Hypotonie, Hypertonie und Tachykardie berichtet.
Allergisch
Hautausschlag, Ödeme, Photosensibilisierung und Pruritus sind gelegentlich aufgetreten.
Hämatologisch
Bei einigen wenigen Patienten wurde über Eosinophilie berichtet. Gelegentlich wurde über eine Knochenmarkdepression berichtet, die sich als Agranulozytose, Leukopenie, Thrombozytopenie und Purpura manifestierte.
Magen-Darm
Übelkeit, Erbrechen, Verdauungsstörungen, Geschmacksstörungen, Durchfall, Anorexie und aphthöse Stomatitis wurden berichtet. (Sehen Anticholinerge Wirkungen .)
Endokrine
Erhöhte oder verminderte Libido, Hodenschwellung, Gynäkomastie bei Männern, Brustvergrößerung und Galaktorrhoe bei Frauen, Erhöhung oder Senkung des Blutzuckerspiegels und Syndrom der unangemessenen ADH-Sekretion wurden bei trizyklischer Verabreichung berichtet.
Sonstiges
Schwindel, Tinnitus, Gewichtszunahme, Schwitzen, Schüttelfrost, Müdigkeit, Schwäche, Hitzegefühl, Gelbsucht, Alopezie, Kopfschmerzen, Verschlimmerung von Asthma, Engwinkelglaukom, Mydriasis und Hyperpyrexie (in Verbindung mit Chlorpromazin) wurden gelegentlich als Nebenwirkungen beobachtet.
Entzugserscheinungen
Die Möglichkeit des Auftretens von Entzugserscheinungen bei abrupter Beendigung der Behandlung nach längerer Verabreichung von SINEQUAN sollte berücksichtigt werden. Diese sind kein Hinweis auf eine Sucht und ein allmähliches Absetzen von Medikamenten sollte diese Symptome nicht verursachen.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Arzneimittel, die von P450 2D6 metabolisiert werden
Die biochemische Aktivität des medikamentenmetabolisierenden Isozyms Cytochrom P450 2D6 (Debrisoquin-Hydroxylase) ist in einer Untergruppe der kaukasischen Bevölkerung reduziert (ca. 7–10 % der Kaukasier sind sogenannte „poor metabolizers“); Zuverlässige Schätzungen der Prävalenz von reduzierter P450-2D6-Isozym-Aktivität bei asiatischen, afrikanischen und anderen Bevölkerungsgruppen sind noch nicht verfügbar. Langsame Metabolisierer haben höhere als erwartete Plasmakonzentrationen von trizyklischen Antidepressiva (TCAs), wenn sie übliche Dosen erhalten. Abhängig von der durch P450 2D6 metabolisierten Fraktion des Arzneimittels kann der Anstieg der Plasmakonzentration gering oder ziemlich groß sein (8-facher Anstieg der Plasma-AUC des TCA).
Darüber hinaus hemmen bestimmte Arzneimittel die Aktivität dieses Isozyms und lassen normale Metabolisierer langsamen Metabolisierern ähneln. Eine Person, die mit einer gegebenen TCA-Dosis stabil ist, kann abrupt toxisch werden, wenn sie eines dieser hemmenden Arzneimittel als Begleittherapie erhält. Zu den Arzneimitteln, die Cytochrom P450 2D6 hemmen, gehören einige, die nicht durch das Enzym verstoffwechselt werden (Chinidin; Cimetidin), und viele, die Substrate für P450 2D6 sind (viele andere Antidepressiva, Phenothiazine und die Typ-1C-Antiarrhythmika Propafenon und Flecainid). Während alle selektiven Serotonin-Wiederaufnahmehemmer (SSRIs), z. B. Citalopram, Escitalopram, Fluoxetin, Sertralin und Paroxetin, P450 2D6 hemmen, können sie im Ausmaß der Hemmung variieren. Das Ausmaß, in dem SSRI-TCA-Wechselwirkungen klinische Probleme aufwerfen können, hängt vom Grad der Hemmung und der Pharmakokinetik der beteiligten SSRI ab. Dennoch ist bei der gleichzeitigen Verabreichung von TCAs mit einem der SSRIs und auch beim Wechsel von einer Klasse zur anderen Vorsicht geboten. Besonders wichtig ist, dass bei einem Patienten, der Fluoxetin abgesetzt wird, angesichts der langen Halbwertszeit des Ausgangsstoffs und des aktiven Metaboliten ausreichend Zeit verstreichen muss, bevor mit der Behandlung mit TCA begonnen wird (mindestens 5 Wochen können erforderlich sein).
Die gleichzeitige Anwendung von trizyklischen Antidepressiva mit Arzneimitteln, die Cytochrom P450 2D6 hemmen können, kann niedrigere Dosen als die üblicherweise verschriebenen entweder für das trizyklische Antidepressivum oder das andere Arzneimittel erfordern. Darüber hinaus kann immer dann, wenn eines dieser anderen Arzneimittel aus der Co-Therapie abgesetzt wird, eine erhöhte Dosis des trizyklischen Antidepressivums erforderlich sein. Es ist wünschenswert, die TCA-Plasmaspiegel zu überwachen, wann immer ein TCA zusammen mit einem anderen Medikament verabreicht wird, von dem bekannt ist, dass es ein Inhibitor von P450 2D6 ist.
Doxepin wird hauptsächlich durch CYP2D6 metabolisiert (mit CYP1A2 und CYP3A4 als Nebenwegen). Inhibitoren oder Substrate von CYP2D6 (dh Chinidin, selektive Serotonin-Wiederaufnahmehemmer [SSRIs]) können bei gleichzeitiger Anwendung die Plasmakonzentration von Doxepin erhöhen. Das Ausmaß der Wechselwirkung hängt von der Variabilität der Wirkung auf CYP2D6 ab. Die klinische Bedeutung dieser Wechselwirkung mit Doxepin wurde nicht systematisch untersucht.
MAO-Hemmer
Bei gleichzeitiger Anwendung bestimmter Arzneimittel mit MAO-Hemmern wurden schwerwiegende Nebenwirkungen und sogar Todesfälle berichtet. Daher sollten MAO-Hemmer mindestens zwei Wochen vor dem vorsichtigen Beginn einer Therapie mit SINEQUAN abgesetzt werden. Die genaue Zeitdauer kann variieren und hängt von dem jeweils verwendeten MAO-Hemmer, der Verabreichungsdauer und der beteiligten Dosierung ab.
Cimetidin
Es wurde berichtet, dass Cimetidin klinisch signifikante Schwankungen der Steady-State-Serumkonzentrationen verschiedener trizyklischer Antidepressiva hervorruft. Schwerwiegende anticholinerge Symptome (dh schwere Mundtrockenheit, Harnverhaltung und verschwommenes Sehen) wurden mit Erhöhungen der Serumspiegel von trizyklischen Antidepressiva zu Beginn der Cimetidin-Therapie in Verbindung gebracht. Darüber hinaus wurden bei Patienten, die bereits Cimetidin einnahmen, höhere Konzentrationen trizyklischer Antidepressiva als erwartet beobachtet, wenn sie damit begonnen wurden. Bei Patienten, von denen berichtet wurde, dass sie mit trizyklischen Antidepressiva unter gleichzeitiger Cimetidin-Therapie gut eingestellt sind, wurde berichtet, dass das Absetzen von Cimetidin die etablierten Steady-State-Serumspiegel trizyklischer Antidepressiva senkt und ihre therapeutischen Wirkungen beeinträchtigt.
Alkohol: Es sollte bedacht werden, dass die Einnahme von Alkohol die Gefahr einer beabsichtigten oder unbeabsichtigten Überdosierung von SINEQUAN erhöhen kann. Dies ist besonders wichtig bei Patienten, die möglicherweise übermäßig Alkohol konsumieren.
Tolazamid
Ein Fall von schwerer Hypoglykämie wurde bei einem Patienten mit Typ-II-Diabetes berichtet, der 11 Tage nach der Zugabe von Doxepin (75 mg/Tag) mit Tolazamid (1 g/Tag) behandelt wurde.
WARNUNGEN
Klinische Verschlechterung und Suizidrisiko
Bei Patienten mit schwerer depressiver Störung (MDD), sowohl bei Erwachsenen als auch bei Kindern, kann es zu einer Verschlechterung ihrer Depression und/oder zum Auftreten von Suizidgedanken und -verhalten (Suizidalität) oder zu ungewöhnlichen Verhaltensänderungen kommen, unabhängig davon, ob sie Antidepressiva einnehmen oder nicht Das Risiko kann bestehen bleiben, bis eine signifikante Remission eintritt. Selbstmord ist ein bekanntes Risiko für Depressionen und bestimmte andere psychiatrische Störungen, und diese Störungen selbst sind die stärksten Prädiktoren für Selbstmord. Es besteht jedoch seit langem die Sorge, dass Antidepressiva bei bestimmten Patienten in den frühen Phasen der Behandlung eine Rolle bei der Induktion einer Verschlechterung der Depression und dem Auftreten von Suizidalität spielen könnten. Gepoolte Analysen von Placebo-kontrollierten Kurzzeitstudien mit Antidepressiva (SSRIs und andere) zeigten, dass diese Medikamente das Risiko von Suizidgedanken und -verhalten (Suizidalität) bei Kindern, Jugendlichen und jungen Erwachsenen (Alter 18-24) mit Major Depression erhöhen Störung (MDD) und andere psychiatrische Störungen. Kurzzeitstudien zeigten bei Erwachsenen über 24 Jahren keine Erhöhung des Suizidalitätsrisikos mit Antidepressiva im Vergleich zu Placebo; bei Erwachsenen ab 65 Jahren kam es unter Antidepressiva im Vergleich zu Placebo zu einer Reduktion.
Die gepoolten Analysen placebokontrollierter Studien bei Kindern und Jugendlichen mit MDD, Zwangsstörungen (OCD) oder anderen psychiatrischen Erkrankungen umfassten insgesamt 24 Kurzzeitstudien mit 9 Antidepressiva bei über 4400 Patienten. Die gepoolten Analysen placebokontrollierter Studien bei Erwachsenen mit MDD oder anderen psychiatrischen Erkrankungen umfassten insgesamt 295 Kurzzeitstudien (mediane Dauer von 2 Monaten) mit 11 Antidepressiva bei über 77.000 Patienten. Es gab beträchtliche Schwankungen des Suizidalitätsrisikos zwischen den Medikamenten, aber bei fast allen untersuchten Medikamenten eine Tendenz zu einem Anstieg bei den jüngeren Patienten. Es gab Unterschiede im absoluten Suizidalitätsrisiko zwischen den verschiedenen Indikationen, mit der höchsten Inzidenz bei MDD. Die Risikounterschiede (Medikament vs. Placebo) waren jedoch innerhalb der Altersschichten und über Indikationen hinweg relativ stabil. Diese Risikounterschiede (Arzneimittel-Placebo-Unterschied in der Anzahl der Fälle von Suizidalität pro 1000 behandelten Patienten) sind in Tabelle 1 aufgeführt.
In keiner der pädiatrischen Studien kam es zu Suiziden. In den Studien mit Erwachsenen gab es Suizide, aber die Anzahl reichte nicht aus, um zu einer Schlussfolgerung über die Wirkung des Medikaments auf Suizide zu gelangen.
Es ist nicht bekannt, ob sich das Suizidrisiko auf eine längerfristige Anwendung erstreckt, dh über mehrere Monate hinaus. Es gibt jedoch erhebliche Hinweise aus placebokontrollierten Erhaltungsstudien bei Erwachsenen mit Depressionen, dass die Anwendung von Antidepressiva das Wiederauftreten von Depressionen verzögern kann.
Alle Patienten, die mit Antidepressiva aus beliebigen Indikationen behandelt werden, sollten angemessen überwacht und engmaschig auf eine klinische Verschlechterung, Suizidalität und ungewöhnliche Verhaltensänderungen beobachtet werden, insbesondere während der ersten Monate einer medikamentösen Therapie oder bei Dosisänderungen oder -steigerungen oder abnimmt.
Die folgenden Symptome, Angst, Agitiertheit, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Ruhelosigkeit), Hypomanie und Manie, wurden bei erwachsenen und pädiatrischen Patienten berichtet, die ebenfalls mit Antidepressiva wegen schwerer depressiver Störung behandelt wurden wie für andere Indikationen, sowohl psychiatrische als auch nichtpsychiatrische. Obwohl ein kausaler Zusammenhang zwischen dem Auftreten solcher Symptome und entweder der Verschlechterung einer Depression und/oder dem Auftreten suizidaler Impulse nicht hergestellt werden konnte, besteht die Sorge, dass solche Symptome Vorboten einer aufkommenden Suizidalität sein könnten.
Bei Patienten, deren Depression sich anhaltend verschlimmert oder die an Suizidalität oder Symptomen leiden, die Vorboten einer Verschlechterung der Depression oder Suizidalität sein könnten, sollte eine Änderung des therapeutischen Schemas, einschließlich eines möglichen Absetzens der Medikation, in Betracht gezogen werden, insbesondere wenn diese Symptome schwerwiegend und abrupt sind zu Beginn oder waren nicht Teil der Symptome des Patienten.
Familien und Betreuer von Patienten, die wegen einer schweren depressiven Störung oder anderen Indikationen, sowohl psychiatrischen als auch nichtpsychiatrischen, mit Antidepressiva behandelt werden, sollten auf die Notwendigkeit aufmerksam gemacht werden, Patienten auf das Auftreten von Unruhe, Reizbarkeit, ungewöhnlichen Verhaltensänderungen und den anderen oben beschriebenen Symptomen zu überwachen , sowie das Auftreten von Suizidalität, und solche Symptome unverzüglich den Gesundheitsdienstleistern zu melden. Eine solche Überwachung sollte die tägliche Beobachtung durch Familien und Betreuer umfassen. Verschreibungen für Sinequan sollten für die kleinste Tablettenmenge im Einklang mit einem guten Patientenmanagement ausgestellt werden, um das Risiko einer Überdosierung zu verringern.
Screening von Patienten auf bipolare Störung
Eine schwere depressive Episode kann die anfängliche Präsentation einer bipolaren Störung sein. Es wird allgemein angenommen (obwohl dies nicht in kontrollierten Studien nachgewiesen wurde), dass die Behandlung einer solchen Episode mit einem Antidepressivum allein die Wahrscheinlichkeit des Auftretens einer gemischten/manischen Episode bei Patienten mit einem Risiko für eine bipolare Störung erhöhen kann. Ob eines der oben beschriebenen Symptome eine solche Konversion darstellt, ist unbekannt. Vor Beginn der Behandlung mit einem Antidepressivum sollten Patienten mit depressiven Symptomen jedoch angemessen untersucht werden, um festzustellen, ob bei ihnen ein Risiko für eine bipolare Störung besteht; Ein solches Screening sollte eine detaillierte psychiatrische Vorgeschichte umfassen, einschließlich einer Familienanamnese von Selbstmord, bipolarer Störung und Depression. Es sollte beachtet werden, dass Sinequan 75 mg nicht zur Behandlung von bipolarer Depression zugelassen ist.
Engwinkelglaukom
Die Pupillenerweiterung, die nach der Anwendung vieler Antidepressiva, einschließlich Sinequan, auftritt, kann bei einem Patienten mit anatomisch engen Winkeln, der keine offene Iridektomie hat, eine Winkelverschlussattacke auslösen.
Verwendung in der Geriatrie: Die Anwendung von SINEQUAN 10 mg in einem einmal täglichen Dosierungsschema bei geriatrischen Patienten sollte sorgfältig an den Zustand des Patienten angepasst werden (siehe VORSICHTSMASSNAHMEN - Geriatrische Verwendung ).
Anwendung in der Schwangerschaft: Reproduktionsstudien wurden an Ratten, Kaninchen, Affen und Hunden durchgeführt und es gab keine Hinweise auf eine Schädigung des Tierfötus. Die Relevanz für den Menschen ist nicht bekannt. Da keine Erfahrungen mit schwangeren Frauen vorliegen, die dieses Arzneimittel erhalten haben, wurde die Sicherheit in der Schwangerschaft nicht nachgewiesen. Bei einem gestillten Säugling, dessen Mutter SINEQUAN einnahm, wurde über Apnoe und Schläfrigkeit berichtet.
Anwendung bei Kindern: Die Anwendung von SINEQUAN 75 mg bei Kindern unter 12 Jahren wird nicht empfohlen, da keine sicheren Bedingungen für seine Anwendung ermittelt wurden.
VORSICHTSMASSNAHMEN
Informationen für Patienten
Verschreibende Ärzte oder andere Angehörige der Gesundheitsberufe sollten Patienten, ihre Familien und ihre Betreuer über die Vorteile und Risiken einer Behandlung mit Sinequan informieren und sie bezüglich der angemessenen Anwendung beraten. Ein Patient Leitfaden für Medikamente über „Antidepressiva, Depressionen und andere schwere psychische Erkrankungen sowie Selbstmordgedanken oder -handlungen“ ist für Sinequan verfügbar. Der verschreibende Arzt oder medizinisches Fachpersonal sollte Patienten, ihre Familien und ihre Betreuer anweisen, die zu lesen Leitfaden für Medikamente und soll ihnen helfen, den Inhalt zu verstehen. Den Patienten sollte die Möglichkeit gegeben werden, den Inhalt des zu diskutieren Leitfaden für Medikamente und um Antworten auf eventuelle Fragen zu erhalten. Der vollständige Text der Leitfaden für Medikamente ist am Ende dieses Dokuments abgedruckt.
Die Patienten sollten auf die folgenden Probleme hingewiesen und gebeten werden, ihren verschreibenden Arzt zu informieren, wenn diese während der Einnahme von Sinequan auftreten.
Klinische Verschlechterung und Suizidrisiko
Patienten, ihre Familien und ihre Betreuer sollten ermutigt werden, auf das Auftreten von Angstzuständen, Unruhe, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Unruhe), Hypomanie, Manie und anderen ungewöhnlichen Verhaltensänderungen zu achten , Verschlechterung von Depressionen und Suizidgedanken, besonders früh während der Behandlung mit Antidepressiva und wenn die Dosis nach oben oder unten angepasst wird. Familien und Betreuer von Patienten sollten angewiesen werden, täglich auf das Auftreten solcher Symptome zu achten, da Änderungen abrupt sein können. Solche Symptome sollten dem verschreibenden Arzt oder medizinischen Fachpersonal des Patienten gemeldet werden, insbesondere wenn sie schwerwiegend sind, abrupt einsetzen oder nicht zu den Symptomen des Patienten gehörten. Symptome wie diese können mit einem erhöhten Risiko für Suizidgedanken und -verhalten verbunden sein und weisen auf die Notwendigkeit einer sehr engmaschigen Überwachung und möglicherweise einer Änderung der Medikation hin.
Die Patienten sollten darauf hingewiesen werden, dass die Einnahme von Sinequan eine leichte Pupillenerweiterung verursachen kann, die bei empfindlichen Personen zu einer Episode eines Engwinkelglaukoms führen kann. Ein vorbestehendes Glaukom ist fast immer ein Offenwinkelglaukom, da ein Winkelblockglaukom, wenn es diagnostiziert wird, definitiv mit einer Iridektomie behandelt werden kann. Das Offenwinkelglaukom ist kein Risikofaktor für das Engwinkelglaukom. Patienten möchten möglicherweise untersucht werden, um festzustellen, ob sie für Winkelverschluss anfällig sind, und sich einem prophylaktischen Verfahren (z. B. Iridektomie) unterziehen, wenn sie anfällig sind.
Pädiatrische Verwendung
Sicherheit und Wirksamkeit in der pädiatrischen Population wurden nicht nachgewiesen (siehe KASTENWARNUNG und WARNUNGEN - Klinische Verschlechterung und Suizidrisiko ).
Jeder, der die Anwendung von SINEQUAN bei einem Kind oder Jugendlichen in Betracht zieht, muss die potenziellen Risiken mit der klinischen Notwendigkeit abwägen.
Schläfrigkeit
Da bei der Anwendung dieses Arzneimittels Schläfrigkeit auftreten kann, sollten die Patienten vor der Möglichkeit gewarnt und davor gewarnt werden, während der Einnahme des Arzneimittels ein Auto zu fahren oder gefährliche Maschinen zu bedienen. Die Patienten sollten auch darauf hingewiesen werden, dass ihre Reaktion auf Alkohol verstärkt werden kann.
Beruhigungsmittel können bei älteren Menschen zu Verwirrung und Übersedierung führen; ältere Patienten sollten im Allgemeinen mit niedrigen Dosen von SINEQUAN begonnen und engmaschig überwacht werden. (Sehen VORSICHTSMASSNAHMEN - Geriatrische Verwendung .)
Selbstmord
Da Suizid bei jedem depressiven Patienten ein inhärentes Risiko darstellt und so lange bestehen bleiben kann, bis eine signifikante Besserung eingetreten ist, sollten die Patienten zu Beginn der Therapie engmaschig überwacht werden. Rezepte sollten für den kleinstmöglichen Betrag ausgestellt werden.
Psychose
Sollten verstärkte Symptome einer Psychose oder ein Wechsel zu manischen Symptomen auftreten, kann es erforderlich sein, die Dosierung zu reduzieren oder dem Dosierungsschema ein starkes Beruhigungsmittel hinzuzufügen.
Geriatrische Verwendung
Es wurde nicht festgestellt, ob kontrollierte klinische Studien mit SINEQUAN eine ausreichende Anzahl von Probanden im Alter von 65 Jahren und älter umfassten, um einen Unterschied im Ansprechen von jüngeren Probanden festzustellen. Andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt. Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein und normalerweise am unteren Ende des Dosierungsbereichs beginnen, um die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und einer Begleiterkrankung oder einer anderen medikamentösen Therapie widerzuspiegeln.
Das Ausmaß der renalen Ausscheidung von SINEQUAN wurde nicht bestimmt. Da bei älteren Patienten mit größerer Wahrscheinlichkeit eine eingeschränkte Nierenfunktion vorliegt, sollte bei der Dosisauswahl sorgfältig vorgegangen werden.
Beruhigungsmittel können bei älteren Menschen zu Verwirrung und Übersedierung führen; ältere Patienten sollten im Allgemeinen mit niedrigen Dosen von SINEQUAN begonnen und engmaschig überwacht werden. (Sehen WARNUNGEN .)
ÜBERDOSIS
Todesfälle können durch Überdosierung mit dieser Klasse von Medikamenten auftreten. Die Einnahme mehrerer Arzneimittel (einschließlich Alkohol) ist bei einer absichtlichen Überdosierung trizyklischer Antidepressiva üblich. Da die Behandlung komplex ist und sich ändert, wird empfohlen, dass sich der Arzt an eine Giftinformationszentrale wendet, um aktuelle Informationen zur Behandlung zu erhalten. Anzeichen und Symptome einer Toxizität entwickeln sich schnell nach einer Überdosierung mit trizyklischen Antidepressiva; daher ist eine Krankenhausüberwachung so schnell wie möglich erforderlich.
Manifestationen
Zu den kritischen Manifestationen einer Überdosierung gehören: Herzrhythmusstörungen, schwere Hypotonie, Krämpfe und ZNS-Depression, einschließlich Koma. Veränderungen im Elektrokardiogramm, insbesondere in der QRS-Achse oder -Breite, sind klinisch signifikante Indikatoren für die Toxizität trizyklischer Antidepressiva.
Andere Anzeichen einer Überdosierung können sein: Verwirrtheit, Konzentrationsstörungen, vorübergehende visuelle Halluzinationen, erweiterte Pupillen, Unruhe, hyperaktive Reflexe, Benommenheit, Benommenheit, Muskelstarre, Erbrechen, Hypothermie, Hyperpyrexie oder eines der unten aufgeführten Symptome NEBENWIRKUNGEN .
Es wurden Todesfälle im Zusammenhang mit Überdosierungen von Doxepin berichtet.
Generelle Empfehlungen
Allgemein
Erhalten Sie ein EKG und leiten Sie sofort eine Herzüberwachung ein. Schützen Sie die Atemwege des Patienten, legen Sie einen intravenösen Zugang an und leiten Sie eine Magendekontamination ein. Eine mindestens sechsstündige Beobachtung mit kardialer Überwachung und Beobachtung auf Anzeichen von ZNS- oder Atemdepression, Hypotonie, Herzrhythmusstörungen und/oder Reizleitungsblockaden und Krampfanfällen wird dringend empfohlen. Wenn zu irgendeinem Zeitpunkt während dieses Zeitraums Anzeichen einer Toxizität auftreten, wird eine erweiterte Überwachung empfohlen. Es gibt Fallberichte von Patienten, die spät nach einer Überdosierung tödlichen Rhythmusstörungen erlagen; Diese Patienten hatten klinische Anzeichen einer signifikanten Vergiftung vor dem Tod und die meisten erhielten eine unzureichende Magen-Darm-Dekontamination. Die Überwachung der Arzneimittelspiegel im Plasma sollte nicht das Management des Patienten leiten.
Magen-Darm-Dekontamination
Alle Patienten mit Verdacht auf eine Überdosierung trizyklischer Antidepressiva sollten eine gastrointestinale Dekontamination erhalten. Dies sollte eine großvolumige Magenspülung gefolgt von Aktivkohle umfassen. Bei Bewusstseinsstörungen sollten die Atemwege vor der Spülung gesichert werden. Erbrechen ist kontraindiziert.
Herz-Kreislauf
Eine maximale Extremitäten-Ableitungs-QRS-Dauer von ≥ 0,10 Sekunden kann der beste Hinweis auf die Schwere der Überdosierung sein. Natriumbicarbonat sollte intravenös verabreicht werden, um den Serum-pH-Wert im Bereich von 7,45 bis 7,55 zu halten. Wenn die pH-Reaktion unzureichend ist, kann auch Hyperventilation verwendet werden. Die gleichzeitige Anwendung von Hyperventilation und Natriumbikarbonat sollte mit äußerster Vorsicht und häufiger pH-Überwachung erfolgen. Ein pH > 7,60 oder ein pCO2
In seltenen Fällen kann die Hämoperfusion bei akuter refraktärer kardiovaskulärer Instabilität bei Patienten mit akuter Toxizität von Vorteil sein. Allerdings wurde berichtet, dass Hämodialyse, Peritonealdialyse, Austauschtransfusionen und forcierte Diurese im Allgemeinen bei Vergiftungen mit trizyklischen Antidepressiva unwirksam sind.
ZNS
Bei Patienten mit ZNS-Depression wird wegen der Möglichkeit einer abrupten Verschlechterung eine frühzeitige Intubation empfohlen. Krampfanfälle sollten mit Benzodiazepinen oder, falls diese unwirksam sind, anderen Antikonvulsiva (z. B. Phenobarbital, Phenytoin) kontrolliert werden. Physostigmin wird nicht empfohlen, außer zur Behandlung lebensbedrohlicher Symptome, die auf andere Therapien nicht angesprochen haben, und dann nur in Absprache mit einem Giftnotrufzentrum.
Psychiatrische Nachsorge
Da eine Überdosierung oft vorsätzlich erfolgt, können Patienten während der Genesungsphase einen Suizidversuch auf andere Weise unternehmen. Eine psychiatrische Überweisung kann angebracht sein.
Pädiatrisches Management
Die Grundsätze der Behandlung von Überdosierungen bei Kindern und Erwachsenen sind ähnlich. Es wird dringend empfohlen, dass sich der Arzt für eine spezifische pädiatrische Behandlung an das örtliche Giftinformationszentrum wendet.
KONTRAINDIKATIONEN
SINEQUAN ist kontraindiziert bei Personen, die eine Überempfindlichkeit gegen das Medikament gezeigt haben. Die Möglichkeit einer Kreuzempfindlichkeit mit anderen Dibenzoxepinen sollte beachtet werden.
SINEQUAN 75 mg ist kontraindiziert bei Patienten mit Glaukom oder einer Neigung zum Harnverhalt. Diese Erkrankungen sollten insbesondere bei älteren Patienten ausgeschlossen werden.
KLINISCHE PHARMAKOLOGIE
Aktionen
Der Wirkungsmechanismus von SINEQUAN (Doxepin-HCl) ist nicht eindeutig bekannt. Es ist weder ein Stimulans des zentralen Nervensystems noch ein Monoaminoxidase-Hemmer. Die aktuelle Hypothese ist, dass die klinischen Effekte zumindest teilweise auf Beeinflussungen der adrenergen Aktivität an den Synapsen zurückzuführen sind, so dass eine Deaktivierung von Norepinephrin durch Wiederaufnahme in die Nervenenden verhindert wird. Tierversuche deuten darauf hin, dass Doxepin-HCl die blutdrucksenkende Wirkung von Guanethidin nicht nennenswert antagonisiert. In Tierstudien wurden anticholinerge, antiserotonin- und antihistaminische Wirkungen auf die glatte Muskulatur nachgewiesen. Bei höheren als den üblichen klinischen Dosen wurde die Norepinephrin-Reaktion bei Tieren verstärkt. Beim Menschen wurde dieser Effekt nicht nachgewiesen.
Bei klinischen Dosierungen von bis zu 150 mg pro Tag kann SINEQUAN 25 mg einem Menschen gleichzeitig mit Guanethidin und verwandten Verbindungen verabreicht werden, ohne die blutdrucksenkende Wirkung zu blockieren. Bei Dosierungen über 150 mg pro Tag wurde über eine Blockierung der blutdrucksenkenden Wirkung dieser Verbindungen berichtet.
SINEQUAN 10mg ist praktisch frei von Euphorie als Nebenwirkung. Charakteristisch für diese Art von Verbindung, SINEQUAN hat nicht gezeigt, dass es die physische Toleranz oder psychische Abhängigkeit hervorruft, die mit süchtig machenden Verbindungen verbunden ist.
INFORMATIONEN ZUM PATIENTEN
Antidepressiva, Depressionen und andere schwere psychische Erkrankungen sowie Selbstmordgedanken oder -handlungen
Lesen Sie den Medikationsleitfaden, der mit dem Antidepressivum von Ihnen oder Ihrem Familienmitglied geliefert wird. In diesem Medikationsleitfaden geht es nur um das Risiko von Selbstmordgedanken und -handlungen mit Antidepressiva.
Sprechen Sie mit Ihrem Gesundheitsdienstleister oder dem Ihres Familienmitglieds über:
Was sind die wichtigsten Informationen, die ich über Antidepressiva, Depressionen und andere schwere psychische Erkrankungen sowie Selbstmordgedanken oder -handlungen wissen sollte?
Rufen Sie sofort einen Arzt an, wenn Sie oder Ihr Familienmitglied eines der folgenden Symptome haben, insbesondere wenn sie neu oder schlimmer sind oder Sie beunruhigen:
Nur einige Menschen sind für diese Probleme gefährdet. Möglicherweise möchten Sie sich einer Augenuntersuchung unterziehen, um festzustellen, ob Sie gefährdet sind, und eine vorbeugende Behandlung erhalten, wenn dies der Fall ist.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Was muss ich sonst noch über Antidepressiva wissen?
Dieser Medikationsleitfaden wurde von der US Food and Drug Administration für alle Antidepressiva genehmigt.
Was ist Singulair und wie wird es angewendet?
Singulair 5 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Asthma, belastungsinduziertem Bronchospasmus und allergischer oder ganzjähriger (ganzjähriger) Rhinitis. Singulair kann allein oder zusammen mit anderen Medikamenten verwendet werden.
Welche Nebenwirkungen kann Singulair haben?
Singulair kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Singulair gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Singulair. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
Montelukast-Natrium, der Wirkstoff in SINGULAIR 4 mg, ist ein selektiver und oral wirksamer Leukotrien-Rezeptorantagonist, der den Cysteinyl-Leukotrien-CysLT1-Rezeptor hemmt.
Montelukast-Natrium wird chemisch als [R-(E)]-1-[[[1-[3-[2-(7-Chlor-2-chinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy) beschrieben -1-Methylethyl)phenyl]propyl]thio]methyl]cyclopropanessigsäure, Mononatriumsalz.
Die Summenformel ist C35H35ClNNaO3S und sein Molekulargewicht beträgt 608,18. Die Strukturformel lautet:
Montelukast-Natrium ist ein hygroskopisches, optisch aktives, weißes bis cremefarbenes Pulver. Montelukast-Natrium ist frei löslich in Ethanol, Methanol und Wasser und praktisch unlöslich in Acetonitril.
Jede 10-mg-Filmtablette von SINGULAIR enthält 10,4 mg Montelukast-Natrium, was 10 mg Montelukast entspricht, und die folgenden Hilfsstoffe: mikrokristalline Cellulose, Lactose-Monohydrat, Croscarmellose-Natrium, Hydroxypropylcellulose und Magnesiumstearat. Die Filmbeschichtung besteht aus Hydroxypropylmethylcellulose, Hydroxypropylcellulose, Titandioxid, rotem Eisenoxid, gelbem Eisenoxid und Carnaubawachs.
Jede 4-mg- und 5-mg-Kautablette SINGULAIR enthält 4,2 bzw. 5,2 mg Montelukast-Natrium, was 4 bzw. 5 mg Montelukast entspricht. Beide Kautabletten enthalten die folgenden Hilfsstoffe: Mannitol, mikrokristalline Cellulose, Hydroxypropylcellulose, rotes Eisenoxid, Croscarmellose-Natrium, Kirscharoma, Aspartam und Magnesiumstearat.
Jede Packung SINGULAIR 4 mg Granulat zum Einnehmen enthält 4,2 mg Montelukast-Natrium, was 4 mg Montelukast entspricht. Die orale Granulatformulierung enthält die folgenden inaktiven Bestandteile: Mannit, Hydroxypropylcellulose und Magnesiumstearat.
INDIKATIONEN
Asthma
SINGULAIR® ist angezeigt zur Prophylaxe und chronischen Behandlung von Asthma bei Erwachsenen und Kindern ab 12 Monaten.
Belastungsinduzierte Bronchokonstriktion (EIB)
SINGULAIR ist angezeigt zur Vorbeugung einer anstrengungsinduzierten Bronchokonstriktion (EIB) bei Patienten ab 6 Jahren.
Allergischer Schnupfen
SINGULAIR 10 mg ist indiziert zur Linderung der Symptome der saisonalen allergischen Rhinitis bei Patienten ab 2 Jahren und der ganzjährigen allergischen Rhinitis bei Patienten ab 6 Monaten. Da der Nutzen von SINGULAIR 4 mg das Risiko neuropsychiatrischer Symptome bei Patienten mit allergischer Rhinitis möglicherweise nicht überwiegt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ], sollte Patienten vorbehalten bleiben, die auf alternative Therapien unzureichend ansprechen oder diese nicht vertragen.
Nutzungsbeschränkungen
SINGULAIR ist nicht angezeigt zur Behandlung eines akuten Asthmaanfalls.
DOSIERUNG UND ANWENDUNG
Asthma
Bei Asthma SINGULAIR einmal täglich abends mit oder ohne Nahrung oral verabreichen. Es wurden keine klinischen Studien bei Patienten mit Asthma durchgeführt, um die relative Wirksamkeit einer morgendlichen gegenüber einer abendlichen Dosierung zu bewerten.
Folgende Dosierungen werden empfohlen:
Patienten, die eine Dosis vergessen haben, sollten die nächste Dosis zur gewohnten Zeit einnehmen und nicht 2 Dosen gleichzeitig einnehmen.
Belastungsinduzierte Bronchokonstriktion (EIB)
Zur Vorbeugung von EIB eine Einzeldosis SINGULAIR 10 mg oral mindestens 2 Stunden vor dem Training verabreichen. Folgende Dosierungen werden empfohlen:
Eine zusätzliche Dosis SINGULAIR sollte nicht innerhalb von 24 Stunden nach einer vorherigen Dosis eingenommen werden. Patienten, die SINGULAIR bereits täglich aus einer anderen Indikation (einschließlich chronischem Asthma) einnehmen, sollten keine zusätzliche Dosis einnehmen, um EIB zu verhindern. Allen Patienten sollte ein kurz wirksamer β-Agonist zur Notfallbehandlung zur Verfügung stehen.
Die tägliche Gabe von SINGULAIR zur chronischen Behandlung von Asthma ist nicht darauf ausgelegt, akute Episoden von EIB zu verhindern.
Allergischer Schnupfen
Bei allergischer Rhinitis SINGULAIR 5 mg einmal täglich oral verabreichen, unabhängig vom Zeitpunkt der Nahrungsaufnahme. Der Verabreichungszeitpunkt bei Patienten mit allergischer Rhinitis kann individuell an die Bedürfnisse des Patienten angepasst werden.
Zur Behandlung der Symptome der saisonalen allergischen Rhinitis werden folgende Dosierungen empfohlen:
Tabelle 3: Empfohlene Dosierung bei saisonaler allergischer Rhinitis
Patienten, die eine Dosis vergessen haben, sollten die nächste Dosis zur gewohnten Zeit einnehmen und nicht 2 Dosen gleichzeitig einnehmen.
Asthma und allergische Rhinitis
Patienten mit Asthma und allergischer Rhinitis nur einmal täglich abends eine Dosis SINGULAIR oral verabreichen.
Patienten, die eine Dosis vergessen haben, sollten die nächste Dosis zur gewohnten Zeit einnehmen und nicht 2 Dosen gleichzeitig einnehmen.
Anweisungen zur Verabreichung von oralen Granulaten
SINGULAIR 4 mg Granulat zum Einnehmen kann entweder direkt in den Mund verabreicht werden, in 1 Teelöffel voll (5 ml) kalter oder zimmerwarmer Babynahrung oder Muttermilch aufgelöst oder mit einem Löffel kalter oder zimmerwarmer weicher Nahrung gemischt werden; Basierend auf Stabilitätsstudien sollten nur Apfelmus, Karotten, Reis oder Eiscreme verwendet werden. Die Packung sollte erst geöffnet werden, wenn sie gebrauchsfertig ist. Nach dem Öffnen der Packung muss die volle Dosis (mit oder ohne Mischen mit Säuglingsnahrung, Muttermilch oder Nahrung) innerhalb von 15 Minuten verabreicht werden. Wenn SINGULAIR 4 mg Granulat zum Einnehmen mit Babynahrung, Muttermilch oder Nahrung gemischt wird, darf es nicht für eine spätere Verwendung aufbewahrt werden. Verwerfen Sie alle nicht verwendeten Teile. SINGULAIR Granulat zum Einnehmen ist nicht dazu bestimmt, zur Verabreichung in einer anderen Flüssigkeit als Säuglingsanfangsnahrung oder Muttermilch aufgelöst zu werden. Flüssigkeiten können jedoch nach der Verabreichung eingenommen werden. SINGULAIR Granulat zum Einnehmen kann unabhängig von den Mahlzeiten verabreicht werden.
WIE GELIEFERT
Darreichungsformen und Stärken
Tablets
10 mg, beige, runde, quadratische Filmtabletten mit dem Code MSD 117 auf der einen Seite und SINGULAIR 4 mg auf der anderen Seite.
Kautabletten
5 mg, rosa, rund, bikonvex geformt, mit Code MSD 275 auf der einen Seite und SINGULAIR auf der anderen Seite.
Kautabletten
4 mg, rosa, oval, bikonvex geformt, mit Code MSD 711 auf der einen Seite und SINGULAIR auf der anderen Seite.
Mündliches Granulat
4 mg, weißes Granulat mit 500 mg Nettogewicht, verpackt in einer kindergesicherten Folienpackung.
Lagerung und Handhabung
SINGULAIR 4 mg Granulat zum Einnehmen : weißes Granulat mit 500 mg Nettogewicht, verpackt in einer kindergesicherten Folienpackung.
NDC 0006-3841-30 Gebrauchseinheit Karton mit 30 Packungen.
SINGULAIR 4 mg Tabletten : Rosafarbene, ovale, bikonvex geformte Kautabletten mit Code MSD 711 auf der einen Seite und SINGULAIR 10 mg auf der anderen Seite.
NDC 0006-1711-31 Gebrauchseinheit Polyethylen hoher Dichte (HDPE) Flaschen mit 30 Stück mit einem kindersicheren Verschluss aus Polypropylen, einer Induktionsversiegelung aus Aluminiumfolie und Silikagel-Trockenmittel.
SINGULAIR 5 mg Tabletten Rosafarbene, runde, bikonvex geformte Kautabletten mit dem Code MSD 275 auf der einen Seite und SINGULAIR auf der anderen Seite.
NDC 0006-9275-31 Gebrauchseinheit Polyethylen hoher Dichte (HDPE) Flaschen mit 30 Stück mit einem kindersicheren Verschluss aus Polypropylen, einer Induktionsversiegelung aus Aluminiumfolie und Silikagel-Trockenmittel.
SINGULAIR 10 mg Tabletten : Beige, abgerundete, quadratische Filmtabletten mit dem Code MSD 117 auf der einen Seite und SINGULAIR 4 mg auf der anderen Seite.
NDC 0006-9117-31 Gebrauchseinheit Polyethylen hoher Dichte (HDPE) Flaschen mit 30 Stück mit einem kindergesicherten Verschluss aus Polypropylen, einer Induktionsversiegelung aus Aluminiumfolie und Silikagel-Trockenmittel
NDC 0006-9117-54 Gebrauchseinheit Polyethylen hoher Dichte (HDPE) Flaschen zu 90 mit einem kindergesicherten Verschluss aus Polypropylen, einer Induktionsversiegelung aus Aluminiumfolie und Silikagel-Trockenmittel.
Lagerung
SINGULAIR 4 mg Granulat zum Einnehmen, 4 mg Kautabletten, 5 mg Kautabletten und 10 mg Filmtabletten bei 20 °C bis 25 °C (68 °F bis 77 °F) lagern, Abweichungen bis 15 °C sind zulässig C bis 30 °C (59 °F bis 86 °F) [siehe USP Kontrollierte Raumtemperatur]. Vor Feuchtigkeit und Licht schützen. In der Originalverpackung aufbewahren.
Vertrieb durch: Merck Sharp & Dohme Corp., eine Tochtergesellschaft von MERCK & CO., INC., Whitehouse Station, USA. Überarbeitet: Februar 2021
NEBENWIRKUNGEN
Die folgenden klinisch signifikanten Nebenwirkungen werden an anderer Stelle in der Kennzeichnung beschrieben:
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider. In der folgenden Beschreibung der Erfahrungen aus klinischen Studien werden Nebenwirkungen unabhängig von der Kausalitätsbewertung aufgeführt.
Die häufigsten Nebenwirkungen (Inzidenz ≥ 5 % und mehr als Placebo; aufgelistet in absteigender Häufigkeit) in kontrollierten klinischen Studien waren: Infektion der oberen Atemwege, Fieber, Kopfschmerzen, Pharyngitis, Husten, Bauchschmerzen, Durchfall, Mittelohrentzündung, Influenza, Rhinorrhoe, Sinusitis, Otitis.
Erwachsene und Jugendliche ab 15 Jahren mit Asthma
SINGULAIR 10 mg wurde in klinischen Studien bei etwa 2950 erwachsenen und jugendlichen Patienten ab 15 Jahren auf Sicherheit untersucht. In placebokontrollierten klinischen Studien traten die folgenden Nebenwirkungen, die mit SINGULAIR 4 mg berichtet wurden, bei mindestens 1 % der Patienten und mit einer höheren Inzidenz als bei mit Placebo behandelten Patienten auf:
Die Häufigkeit weniger häufiger Nebenwirkungen war zwischen SINGULAIR und Placebo vergleichbar.
Das Sicherheitsprofil von SINGULAIR 10 mg bei Verabreichung als Einzeldosis zur Vorbeugung von EIB bei erwachsenen und jugendlichen Patienten ab 15 Jahren entsprach dem zuvor für SINGULAIR beschriebenen Sicherheitsprofil.
Insgesamt wurden in klinischen Studien 569 Patienten mindestens 6 Monate, 480 ein Jahr und 49 zwei Jahre mit SINGULAIR 10 mg behandelt. Bei längerer Behandlung änderte sich das Nebenwirkungsprofil nicht signifikant.
Pädiatrische Patienten im Alter von 6 bis 14 Jahren mit Asthma
Die Sicherheit von SINGULAIR 10 mg wurde bei 476 pädiatrischen Patienten im Alter von 6 bis 14 Jahren untersucht. Insgesamt wurden 289 pädiatrische Patienten in klinischen Studien mit SINGULAIR 10 mg für mindestens 6 Monate und 241 für ein Jahr oder länger behandelt. Das Sicherheitsprofil von SINGULAIR in der 8-wöchigen, doppelblinden, pädiatrischen Wirksamkeitsstudie war im Allgemeinen ähnlich dem Sicherheitsprofil für Erwachsene. Bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren, die SINGULAIR 5 mg erhielten, traten die folgenden Reaktionen mit einer Häufigkeit von ≥ 2 % und häufiger auf als bei pädiatrischen Patienten, die Placebo erhielten: Pharyngitis, Grippe, Fieber, Sinusitis, Übelkeit, Durchfall, Dyspepsie, Otitis, Virusinfektion und Laryngitis. Die Häufigkeit weniger häufiger Nebenwirkungen war zwischen SINGULAIR 4 mg und Placebo vergleichbar. Bei längerer Behandlung änderte sich das Nebenwirkungsprofil nicht signifikant.
Das Sicherheitsprofil von SINGULAIR 4 mg bei Verabreichung als Einzeldosis zur Prävention von EIB bei pädiatrischen Patienten ab 6 Jahren entsprach dem zuvor für SINGULAIR beschriebenen Sicherheitsprofil.
In Studien zur Bewertung der Wachstumsrate stimmte das Sicherheitsprofil bei diesen pädiatrischen Patienten mit dem zuvor für SINGULAIR beschriebenen Sicherheitsprofil überein. In einer 56-wöchigen, doppelblinden Studie zur Untersuchung der Wachstumsrate bei pädiatrischen Patienten im Alter von 6 bis 8 Jahren, die SINGULAIR erhielten, traten die folgenden Reaktionen, die zuvor bei der Anwendung von SINGULAIR in dieser Altersgruppe nicht beobachtet wurden, mit einer Häufigkeit von ≥ 2 % und häufiger auf als bei pädiatrischen Patienten, die Placebo erhielten: Kopfschmerzen, Rhinitis (infektiös), Windpocken, Gastroenteritis, atopische Dermatitis, akute Bronchitis, Zahninfektion, Hautinfektion und Kurzsichtigkeit.
Pädiatrische Patienten im Alter von 2 bis 5 Jahren mit Asthma
Die Sicherheit von SINGULAIR wurde bei 573 pädiatrischen Patienten im Alter von 2 bis 5 Jahren in Einzel- und Mehrfachdosisstudien untersucht. Insgesamt wurden in klinischen Studien 426 pädiatrische Patienten im Alter von 2 bis 5 Jahren mit SINGULAIR 10 mg für mindestens 3 Monate, 230 für 6 Monate oder länger und 63 Patienten für ein Jahr oder länger behandelt. Bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren, die SINGULAIR erhielten, traten die folgenden Reaktionen mit einer Häufigkeit von ≥ 2 % und häufiger auf als bei pädiatrischen Patienten, die Placebo erhielten: Fieber, Husten, Bauchschmerzen, Durchfall, Kopfschmerzen, Schnupfen, Sinusitis, Otitis, Influenza, Hautausschlag, Ohrenschmerzen, Gastroenteritis, Ekzem, Urtikaria, Windpocken, Lungenentzündung, Dermatitis und Konjunktivitis.
Pädiatrische Patienten im Alter von 6 bis 23 Monaten mit Asthma
Sicherheit und Wirksamkeit bei pädiatrischen Asthmapatienten unter 12 Monaten sind nicht erwiesen.
Die Sicherheit von SINGULAIR 4 mg wurde bei 175 pädiatrischen Patienten im Alter von 6 bis 23 Monaten untersucht. Das Sicherheitsprofil von SINGULAIR in einer 6-wöchigen, doppelblinden, placebokontrollierten klinischen Studie war im Allgemeinen ähnlich dem Sicherheitsprofil bei Erwachsenen und pädiatrischen Patienten im Alter von 2 bis 14 Jahren. Bei pädiatrischen Patienten im Alter von 6 bis 23 Monaten, die SINGULAIR 5 mg erhielten, traten die folgenden Reaktionen mit einer Häufigkeit von ≥ 2 % und häufiger auf als bei pädiatrischen Patienten, die Placebo erhielten: Infektion der oberen Atemwege, Keuchen; Mittelohrentzündung; Pharyngitis, Mandelentzündung, Husten; und Schnupfen. Die Häufigkeit weniger häufiger Nebenwirkungen war zwischen SINGULAIR und Placebo vergleichbar.
Erwachsene und Jugendliche ab 15 Jahren mit saisonaler allergischer Rhinitis
SINGULAIR wurde in klinischen Studien an 2199 erwachsenen und jugendlichen Patienten ab 15 Jahren auf Sicherheit untersucht. SINGULAIR 10 mg, das einmal täglich morgens oder abends verabreicht wurde, hatte ein ähnliches Sicherheitsprofil wie Placebo. In placebokontrollierten klinischen Studien wurde die folgende Reaktion mit SINGULAIR 5 mg mit einer Häufigkeit von ≥ 1 % und einer höheren Inzidenz als Placebo berichtet: Infektion der oberen Atemwege, 1,9 % der Patienten, die SINGULAIR 4 mg erhielten, gegenüber 1,5 % der Patienten, die Placebo erhielten. In einer 4-wöchigen placebokontrollierten klinischen Studie stimmte das Sicherheitsprofil mit dem in 2-wöchigen Studien beobachteten überein. Die Inzidenz von Somnolenz war in allen Studien ähnlich wie bei Placebo.
Pädiatrische Patienten im Alter von 2 bis 14 Jahren mit saisonaler allergischer Rhinitis
SINGULAIR 5 mg wurde bei 280 pädiatrischen Patienten im Alter von 2 bis 14 Jahren in einer 2-wöchigen, multizentrischen, doppelblinden, placebokontrollierten Parallelgruppen-Sicherheitsstudie untersucht. SINGULAIR, das einmal täglich abends verabreicht wurde, hatte ein ähnliches Sicherheitsprofil wie Placebo. In dieser Studie traten die folgenden Reaktionen mit einer Häufigkeit von ≥ 2 % und einer höheren Inzidenz als unter Placebo auf: Kopfschmerzen, Mittelohrentzündung, Pharyngitis und Infektion der oberen Atemwege.
Erwachsene und Jugendliche ab 15 Jahren mit ganzjähriger allergischer Rhinitis
Die Sicherheit von SINGULAIR wurde bei 3357 erwachsenen und jugendlichen Patienten ab 15 Jahren mit perennialer allergischer Rhinitis untersucht, von denen 1632 SINGULAIR in zwei 6-wöchigen klinischen Studien erhielten. Einmal täglich verabreichtes SINGULAIR 10 mg hatte ein Sicherheitsprofil, das mit dem bei Patienten mit saisonaler allergischer Rhinitis beobachteten und dem von Placebo vergleichbar war. In diesen beiden Studien wurden die folgenden Reaktionen unter SINGULAIR mit einer Häufigkeit von ≥ 1 % und einer höheren Inzidenz als unter Placebo berichtet: Sinusitis, Infektion der oberen Atemwege, Sinuskopfschmerz, Husten, Nasenbluten und erhöhte ALT. Die Inzidenz von Somnolenz war ähnlich wie bei Placebo.
Pädiatrische Patienten im Alter von 6 Monaten bis 14 Jahren mit ganzjähriger allergischer Rhinitis
Die Sicherheit bei Patienten im Alter von 2 bis 14 Jahren mit ganzjähriger allergischer Rhinitis wird durch die Sicherheit bei Patienten im Alter von 2 bis 14 Jahren mit saisonaler allergischer Rhinitis gestützt. Die Sicherheit bei Patienten im Alter von 6 bis 23 Monaten wird durch Daten aus Pharmakokinetik- und Sicherheits- und Wirksamkeitsstudien bei Asthma in dieser pädiatrischen Population und aus Pharmakokinetikstudien bei Erwachsenen gestützt.
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von SINGULAIR nach der Zulassung festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Erkrankungen des Blutes und des Lymphsystems erhöhte Blutungsneigung, Thrombozytopenie
Störungen des Immunsystems Überempfindlichkeitsreaktionen einschließlich Anaphylaxie, hepatische eosinophile Infiltration
Psychische Störungen einschließlich, aber nicht beschränkt auf Agitation, aggressives Verhalten oder Feindseligkeit, Ängstlichkeit, Depression, Orientierungslosigkeit, Aufmerksamkeitsstörungen, Traumanomalien, Dysphämie (Stottern), Halluzinationen, Schlaflosigkeit, Reizbarkeit, Gedächtnisstörungen, Zwangssymptome, Ruhelosigkeit, Somnambulismus, Suizidgedanken und -verhalten (einschließlich Suizid), Tic und Tremor [vgl Eingerahmte Warnung , WARNUNGEN UND VORSICHTSMASSNAHMEN ]
Erkrankungen des Nervensystems Benommenheit, Parästhesie/Hypästhesie, Krampfanfälle
Herzerkrankungen Herzklopfen
Erkrankungen der Atemwege, des Brustraums und Mediastinums Nasenbluten, pulmonale Eosinophilie
Gastrointestinale Störungen Durchfall, Dyspepsie, Übelkeit, Pankreatitis, Erbrechen
Leber- und Gallenerkrankungen Fälle von cholestatischer Hepatitis, hepatozellulärer Leberschädigung und Leberschädigung gemischten Musters wurden bei mit SINGULAIR behandelten Patienten berichtet. Die meisten davon traten in Kombination mit anderen Störfaktoren auf, wie z. B. der Einnahme anderer Medikamente, oder wenn SINGULAIR Patienten verabreicht wurde, die ein zugrunde liegendes Potenzial für Lebererkrankungen wie Alkoholkonsum oder andere Formen von Hepatitis hatten.
Erkrankungen der Haut und des Unterhautgewebes Angioödem, Blutergüsse, Erythema multiforme, Erythema nodosum, Juckreiz, Stevens-Johnson-Syndrom/toxische epidermale Nekrolyse, Urtikaria
Erkrankungen des Bewegungsapparates und des Bindegewebes Arthralgie, Myalgie einschließlich Muskelkrämpfe
Erkrankungen der Nieren und Harnwege Enuresis bei Kindern
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Ödem
Patienten mit Asthma, die mit SINGULAIR behandelt werden, können eine systemische Eosinophilie aufweisen, die sich manchmal mit klinischen Merkmalen einer Vaskulitis präsentiert, die mit dem Churg-Strauss-Syndrom vereinbar ist, einer Erkrankung, die häufig mit einer systemischen Kortikosteroidtherapie behandelt wird. Diese Reaktionen wurden manchmal mit der Reduzierung der oralen Kortikosteroidtherapie in Verbindung gebracht. Ärzte sollten auf Eosinophilie, vaskulitischen Ausschlag, sich verschlechternde Lungensymptome, kardiale Komplikationen und/oder Neuropathie bei ihren Patienten achten [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Es ist keine Dosisanpassung erforderlich, wenn SINGULAIR 5 mg zusammen mit Theophyllin, Prednison, Prednisolon, oralen Kontrazeptiva, Fexofenadin, Digoxin, Warfarin, Gemfibrozil, Itraconazol, Schilddrüsenhormonen, sedierenden Hypnotika, nichtsteroidalen entzündungshemmenden Mitteln, Benzodiazepinen, Dekongestiva, und Cytochrom P450 (CYP)-Enzyminduktoren [vgl KLINISCHE PHARMAKOLOGIE ].
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Neuropsychiatrische Ereignisse
Bei der Anwendung von SINGULAIR wurde über schwerwiegende neuropsychiatrische (NP) Ereignisse berichtet. Diese Postmarketing-Berichte waren sehr unterschiedlich und umfassten unter anderem Agitiertheit, aggressives Verhalten oder Feindseligkeit, Ängstlichkeit, Depression, Orientierungslosigkeit, Aufmerksamkeitsstörungen, Traumanomalien, Dysphämie (Stottern), Halluzinationen, Schlaflosigkeit, Reizbarkeit, Gedächtnisstörungen, Zwangssymptome, Unruhe, Somnambulismus, Selbstmordgedanken und -verhalten (einschließlich Selbstmord), Tic und Tremor. NP-Ereignisse wurden bei erwachsenen, jugendlichen und pädiatrischen Patienten mit und ohne psychiatrische Erkrankung in der Vorgeschichte berichtet. NP-Ereignisse wurden hauptsächlich während der Behandlung mit SINGULAIR 10 mg berichtet, aber einige wurden nach Absetzen von SINGULAIR 5 mg berichtet. Tierversuche zeigten, dass sich Montelukast bei Ratten im Gehirn verteilt [vgl KLINISCHE PHARMAKOLOGIE ]; Die Mechanismen, die SINGULAIR-assoziierten NP-Ereignissen zugrunde liegen, sind jedoch derzeit nicht gut verstanden. Basierend auf den verfügbaren Daten ist es schwierig, Risikofaktoren für NP-Ereignisse bei der Anwendung von SINGULAIR 10 mg zu identifizieren oder das Risiko zu quantifizieren.
Aufgrund des Risikos von NP-Ereignissen überwiegt der Nutzen von SINGULAIR die Risiken bei manchen Patienten möglicherweise nicht, insbesondere wenn die Krankheitssymptome leicht sind und mit alternativen Therapien angemessen behandelt werden können. Reservieren Sie die Anwendung von SINGULAIR 4 mg für Patienten mit allergischer Rhinitis, die unzureichend auf alternative Therapien ansprechen oder diese nicht vertragen [siehe INDIKATIONEN ]. Bei Patienten mit Asthma oder anstrengungsinduzierter Bronchokonstriktion sollten Sie Nutzen und Risiken abwägen, bevor Sie SINGULAIR verschreiben.
Besprechen Sie Nutzen und Risiken der Anwendung von SINGULAIR 10 mg mit Patienten und Pflegepersonal, wenn Sie SINGULAIR verschreiben. Weisen Sie Patienten und/oder Pflegekräfte an, bei der Einnahme von SINGULAIR auf Verhaltensänderungen oder neue NP-Symptome zu achten. Wenn Verhaltensänderungen beobachtet werden oder wenn neue NP-Symptome oder Suizidgedanken und/oder -verhalten auftreten, raten Sie den Patienten, SINGULAIR 10 mg abzusetzen und sich unverzüglich an einen Arzt zu wenden. In vielen Fällen verschwanden die Symptome nach Beendigung der Behandlung mit SINGULAIR; In einigen Fällen blieben die Symptome jedoch nach Absetzen von SINGULAIR bestehen. Daher weiterhin überwachen und unterstützend behandeln, bis die Symptome abgeklungen sind. Nutzen und Risiken einer Wiederaufnahme der Behandlung mit SINGULAIR 10 mg neu bewerten, wenn solche Ereignisse auftreten.
Akute Asthma
SINGULAIR ist nicht angezeigt zur Anwendung bei der Aufhebung von Bronchospasmen bei akuten Asthmaanfällen, einschließlich Status asthmaticus. Den Patienten sollte geraten werden, geeignete Notfallmedikamente bereitzuhalten. Bei akuten Exazerbationen des Asthmas kann die Therapie mit SINGULAIR 4 mg fortgesetzt werden. Patienten, die nach körperlicher Anstrengung Asthma-Exazerbationen erleiden, sollten zur Notfallbehandlung einen kurz wirksamen inhalativen β-Agonisten zur Verfügung haben.
Gleichzeitige Anwendung von Kortikosteroiden
Während die Dosis eines inhalativen Kortikosteroids unter ärztlicher Aufsicht schrittweise reduziert werden kann, sollte SINGULAIR nicht abrupt durch inhalative oder orale Kortikosteroide ersetzt werden.
Aspirin-Empfindlichkeit
Patienten mit bekannter Aspirin-Empfindlichkeit sollten Aspirin oder nicht-steroidale entzündungshemmende Mittel während der Einnahme von SINGULAIR weiterhin vermeiden. Obwohl SINGULAIR bei der Verbesserung der Atemwegsfunktion bei Asthmatikern mit dokumentierter Aspirin-Empfindlichkeit wirksam ist, wurde nicht gezeigt, dass es die Bronchokonstriktor-Reaktion auf Aspirin und andere nicht-steroidale entzündungshemmende Arzneimittel bei Aspirin-empfindlichen Asthmapatienten verkürzt [siehe Klinische Studien ].
Eosinophile Bedingungen
Patienten mit Asthma, die mit SINGULAIR behandelt werden, können eine systemische Eosinophilie aufweisen, die sich manchmal mit klinischen Merkmalen einer Vaskulitis präsentiert, die mit dem Churg-Strauss-Syndrom vereinbar ist, einer Erkrankung, die häufig mit einer systemischen Kortikosteroidtherapie behandelt wird. Diese Ereignisse wurden manchmal mit einer Reduktion der oralen Kortikosteroidtherapie in Verbindung gebracht. Ärzte sollten bei ihren Patienten auf Eosinophilie, vaskulitischen Hautausschlag, sich verschlechternde Lungensymptome, kardiale Komplikationen und/oder Neuropathie achten. Ein kausaler Zusammenhang zwischen SINGULAIR 10 mg und diesen Grunderkrankungen wurde nicht nachgewiesen [siehe NEBENWIRKUNGEN ].
Risiko bei Patienten mit Phenylketonurie
SINGULAIR 4 mg enthält Aspartam, eine Quelle für Phenylalanin. Phenylalanin kann für Patienten mit Phenylketonurie (PKU) schädlich sein. Jede 4-mg- und 5-mg-Kautablette enthält 0,674 mg bzw. 0,842 mg Phenylalanin. Bevor Sie einem Patienten mit PKU SINGULAIR verschreiben, berücksichtigen Sie die kombinierte tägliche Menge an Phenylalanin aus allen Quellen, einschließlich SINGULAIR.
Informationen zur Patientenberatung
Weisen Sie für die Tabletten und Kautabletten den Patienten und/oder die Pflegekraft an, die von der FDA zugelassene Patientenkennzeichnung zu lesen ( Leitfaden für Medikamente ).
Weisen Sie für das orale Granulat den Patienten und/oder die Pflegekraft an, die von der FDA zugelassene Patientenkennzeichnung zu lesen ( Medikationsleitfaden und Gebrauchsanweisung ).
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
In Kanzerogenitätsstudien über 2 Jahre an Sprague-Dawley-Ratten oder 92 Wochen an Mäusen mit oralen Sondendosen von bis zu 200 mg/kg/Tag bzw. 100 mg/kg/Tag wurden keine Hinweise auf Tumorigenität gefunden. Die geschätzte Exposition bei Ratten betrug etwa das 120- bzw. 75-fache der AUC für Erwachsene bzw. Kinder bei der maximal empfohlenen oralen Tagesdosis. Die geschätzte Exposition bei Mäusen betrug etwa das 45- bzw. 25-fache der AUC für Erwachsene bzw. Kinder bei der maximal empfohlenen oralen Tagesdosis.
Montelukast zeigte in den folgenden Assays keine Hinweise auf mutagene oder klastogene Aktivität: dem mikrobiellen Mutagenese-Assay, dem V-79-Säugerzell-Mutagenese-Assay, dem alkalischen Elutions-Assay in Ratten-Hepatozyten, dem Chromosomenaberrations-Assay in Eierstockzellen des chinesischen Hamsters und im in vivo Knochenmark-Chromosomenaberrationsassay der Maus.
In Fertilitätsstudien an weiblichen Ratten führte Montelukast bei einer oralen Dosis von 200 mg/kg zu einer Verringerung der Fertilitäts- und Fruchtbarkeitsindizes (die geschätzte Exposition betrug etwa das 70-fache der AUC für Erwachsene bei der maximal empfohlenen oralen Tagesdosis). Bei einer oralen Dosis von 100 mg/kg wurden keine Auswirkungen auf die weibliche Fertilität oder Fertilität beobachtet (die geschätzte Exposition betrug etwa das 20-fache der AUC für Erwachsene bei der maximal empfohlenen oralen Tagesdosis). Montelukast hatte bei oralen Dosen von bis zu 800 mg/kg keine Auswirkungen auf die Fertilität männlicher Ratten (die geschätzte Exposition betrug etwa das 160-fache der AUC für Erwachsene bei der maximal empfohlenen oralen Tagesdosis).
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Zusammenfassung der Risiken
Verfügbare Daten aus veröffentlichten prospektiven und retrospektiven Kohortenstudien über Jahrzehnte mit der Anwendung von Montelukast bei schwangeren Frauen haben kein arzneimittelbedingtes Risiko schwerer Geburtsfehler festgestellt [siehe Daten ]. In Reproduktionsstudien an Tieren wurden bei oraler Gabe von Montelukast an trächtige Ratten und Kaninchen während der Organogenese in Dosen, die etwa dem 100- bzw. 110-Fachen der maximal empfohlenen täglichen oralen Dosis beim Menschen (MRHDOD) entsprachen, basierend auf den AUCs, keine nachteiligen Auswirkungen auf die Entwicklung beobachtet [siehe Daten ].
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten für die angegebene Population ist unbekannt. Alle Schwangerschaften haben ein Hintergrundrisiko für Geburtsfehler, Verlust oder andere nachteilige Folgen. In dem
In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwerwiegende Geburtsfehler und Fehlgeburten bei klinisch anerkannten Schwangerschaften bei 2–4 % bzw. 15–20 %.
Klinische Überlegungen
Krankheitsbedingtes maternales und/oder embryo/fetales Risiko
Schlecht oder mäßig kontrolliertes Asthma in der Schwangerschaft erhöht das mütterliche Risiko für perinatale Nebenwirkungen wie Präeklampsie und Frühgeburtlichkeit, niedriges Geburtsgewicht und geringes Gestationsalter.
Daten
Menschliche Daten
Veröffentlichte Daten aus prospektiven und retrospektiven Kohortenstudien haben keinen Zusammenhang mit der Anwendung von SINGULAIR während der Schwangerschaft und schweren Geburtsfehlern festgestellt. Verfügbare Studien weisen methodische Einschränkungen auf, darunter eine kleine Stichprobengröße, teilweise retrospektive Datenerhebung und inkonsistente Vergleichsgruppen.
Tierdaten
In Studien zur embryofetalen Entwicklung verursachte Montelukast, das trächtigen Ratten und Kaninchen während der Organogenese (Tragzeit 6 bis 17 bei Ratten und 6 bis 18 bei Kaninchen) verabreicht wurde, bei maternalen oralen Dosen von bis zu 400 und 300 mg/kg keine nachteiligen Auswirkungen auf die Entwicklung /Tag bei Ratten bzw. Kaninchen (etwa das 100- bzw. 110-fache der AUC beim Menschen bei der MRHDOD).
Stillzeit
Zusammenfassung der Risiken
Eine veröffentlichte klinische Laktationsstudie berichtet über das Vorhandensein von Montelukast in der Muttermilch. Verfügbare Daten zu den Wirkungen des Medikaments auf Säuglinge, entweder direkt [siehe Pädiatrische Verwendung oder durch die Muttermilch, weisen nicht auf ein signifikantes Risiko von Nebenwirkungen durch die Exposition gegenüber SINGULAIR hin. Die Auswirkungen des Medikaments auf die Milchproduktion sind nicht bekannt. Die Entwicklungs- und Gesundheitsvorteile des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an SINGULAIR 5 mg und möglichen Nebenwirkungen auf den gestillten Säugling durch SINGULAIR 10 mg oder aufgrund des zugrunde liegenden Zustands der Mutter berücksichtigt werden.
Pädiatrische Verwendung
Die Sicherheit und Wirksamkeit von SINGULAIR bei Asthma wurde bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren nachgewiesen. Die Anwendung von SINGULAIR 4 mg für diese Indikation wird durch Belege aus gut kontrollierten Studien gestützt. Die Sicherheits- und Wirksamkeitsdaten in dieser Altersgruppe ähneln denen bei Erwachsenen [siehe NEBENWIRKUNGEN , KLINISCHE PHARMAKOLOGIE , Spezifische Populationen , und Klinische Studien ].
Die Wirksamkeit von SINGULAIR zur Behandlung von saisonaler allergischer Rhinitis bei pädiatrischen Patienten im Alter von 2 bis 14 Jahren und zur Behandlung von ganzjähriger allergischer Rhinitis bei pädiatrischen Patienten im Alter von 6 Monaten bis 14 Jahren wurde nachgewiesen und wird durch Extrapolation der nachgewiesenen Wirksamkeit gestützt bei Patienten ab 15 Jahren mit allergischer Rhinitis sowie der Annahme, dass der Krankheitsverlauf, die Pathophysiologie und die Wirkung des Arzneimittels in diesen Populationen im Wesentlichen ähnlich sind.
Die Sicherheit von SINGULAIR 4 mg Kautabletten bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren mit Asthma wurde durch angemessene und gut kontrollierte Daten belegt [siehe NEBENWIRKUNGEN ]. Die Wirksamkeit von SINGULAIR 5 mg in dieser Altersgruppe wird aus der nachgewiesenen Wirksamkeit bei Patienten ab 6 Jahren mit Asthma extrapoliert und basiert auf ähnlichen pharmakokinetischen Daten sowie der Annahme, dass der Krankheitsverlauf, die Pathophysiologie und die Wirkung des Arzneimittels im Wesentlichen ähnlich sind unter diesen Populationen. Die Wirksamkeit in dieser Altersgruppe wird durch explorative Wirksamkeitsbewertungen aus einer großen, gut kontrollierten Sicherheitsstudie gestützt, die an Patienten im Alter von 2 bis 5 Jahren durchgeführt wurde.
Die Sicherheit von SINGULAIR 4 mg oralem Granulat bei pädiatrischen Patienten im Alter von 12 bis 23 Monaten mit Asthma wurde in einer Analyse von 172 pädiatrischen Patienten, von denen 124 mit SINGULAIR behandelt wurden, in einer 6-wöchigen, doppelblinden Placebo-Studie nachgewiesen -kontrollierte Studie [vgl NEBENWIRKUNGEN ]. Die Wirksamkeit von SINGULAIR in dieser Altersgruppe wird aus der nachgewiesenen Wirksamkeit bei Patienten ab 6 Jahren mit Asthma extrapoliert, basierend auf einer ähnlichen mittleren systemischen Exposition (AUC) und der Tatsache, dass der Krankheitsverlauf, die Pathophysiologie und die Wirkung des Arzneimittels bei diesen Populationen im Wesentlichen ähnlich sind , unterstützt durch Wirksamkeitsdaten aus einer Sicherheitsstudie, in der die Wirksamkeit eine explorative Bewertung war.
Die Sicherheit von SINGULAIR 4 mg und 5 mg Kautabletten bei pädiatrischen Patienten im Alter von 2 bis 14 Jahren mit allergischer Rhinitis wird durch Daten aus Studien gestützt, die an pädiatrischen Patienten im Alter von 2 bis 14 Jahren mit Asthma durchgeführt wurden. Eine Sicherheitsstudie bei pädiatrischen Patienten im Alter von 2 bis 14 Jahren mit saisonaler allergischer Rhinitis zeigte ein ähnliches Sicherheitsprofil [siehe NEBENWIRKUNGEN ]. Die Sicherheit von SINGULAIR 4 mg Granulat zum Einnehmen bei pädiatrischen Patienten im Alter von 6 Monaten mit perennialer allergischer Rhinitis wird durch Extrapolation von Sicherheitsdaten gestützt, die aus Studien an pädiatrischen Patienten im Alter von 6 bis 23 Monaten mit Asthma und aus pharmakokinetischen Daten gewonnen wurden Vergleich systemischer Expositionen bei Patienten im Alter von 6 bis 23 Monaten mit systemischen Expositionen bei Erwachsenen.
Die Sicherheit und Wirksamkeit bei pädiatrischen Patienten unter 12 Monaten mit Asthma, unter 6 Monaten mit ganzjähriger allergischer Rhinitis und unter 6 Jahren mit anstrengungsinduzierter Bronchokonstriktion wurde nicht nachgewiesen.
Wachstumsrate bei pädiatrischen Patienten
Eine 56-wöchige, multizentrische, doppelblinde, randomisierte, aktiv- und placebokontrollierte Parallelgruppenstudie wurde durchgeführt, um die Wirkung von SINGULAIR 4 mg auf die Wachstumsrate bei 360 Patienten mit leichtem Asthma im Alter von 6 bis 8 Jahren zu beurteilen. Die Behandlungsgruppen umfassten SINGULAIR 5 mg einmal täglich, Placebo und Beclomethasondipropionat, verabreicht als 168 µg zweimal täglich mit einem Spacer. Für jedes Subjekt wurde eine Wachstumsrate als Steigung einer linearen Regressionslinie definiert, die an die Größenmessungen über 56 Wochen angepasst war. Der primäre Vergleich war der Unterschied in den Wachstumsraten zwischen der SINGULAIR- und der Placebo-Gruppe. Wachstumsraten, ausgedrückt als Mittelwert der kleinsten Quadrate (LS) (95 % KI) in cm/Jahr, für die SINGULAIR-, Placebo- und Beclomethason-Behandlungsgruppen betrugen 5,67 (5,46; 5,88), 5,64 (5,42; 5,86) und 4,86 ( 4,64, 5,08). Die Unterschiede in den Wachstumsraten, ausgedrückt als Mittelwert der kleinsten Quadrate (LS) (95 %-KI) in cm/Jahr, für die Behandlungsgruppen mit SINGULAIR 5 mg minus Placebo, Beclomethason minus Placebo und SINGULAIR 5 mg minus Beclomethason betrugen 0,03 (-0,26; 0,31). , -0,78 (-1,06, -0,49); bzw. 0,81 (0,53, 1,09). Die Wachstumsrate (ausgedrückt als mittlere Veränderung der Körpergröße im Laufe der Zeit) für jede Behandlungsgruppe ist in ABBILDUNG 1 dargestellt.
Abbildung 1: Veränderung der Körpergröße (cm) vom Randomisierungsbesuch bis zur geplanten Woche (Mittelwert der Behandlungsgruppe ± Standardfehler* des Mittelwerts)
Geriatrische Verwendung
Von der Gesamtzahl der Studienteilnehmer in klinischen Studien zu Montelukast waren 3,5 % 65 Jahre und älter und 0,4 % 75 Jahre und älter. Insgesamt wurden keine Unterschiede in der Sicherheit oder Wirksamkeit zwischen diesen Probanden und jüngeren Probanden beobachtet, und andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt, aber eine größere Empfindlichkeit einiger älterer Personen kann nicht ausgeschlossen werden. Das pharmakokinetische Profil und die orale Bioverfügbarkeit einer oralen Einzeldosis von 10 mg Montelukast sind bei älteren und jüngeren Erwachsenen ähnlich. Die Plasmahalbwertszeit von Montelukast ist bei älteren Patienten etwas länger. Bei älteren Patienten ist keine Dosisanpassung erforderlich.
Leberfunktionsstörung
Bei Patienten mit leichter bis mittelschwerer Leberinsuffizienz wird keine Dosisanpassung empfohlen [siehe KLINISCHE PHARMAKOLOGIE ].
Nierenfunktionsstörung
Bei Patienten mit Niereninsuffizienz wird keine Dosisanpassung empfohlen [siehe KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Zur Behandlung einer Überdosierung mit SINGULAIR liegen keine spezifischen Informationen vor. Im Falle einer Überdosierung ist es sinnvoll, die üblichen unterstützenden Maßnahmen anzuwenden; B. nicht resorbiertes Material aus dem Gastrointestinaltrakt entfernen, klinisch überwachen und bei Bedarf eine unterstützende Therapie einleiten. Es ist nicht bekannt, ob Montelukast durch Peritonealdialyse oder Hämodialyse entfernt wird.
KONTRAINDIKATIONEN
SINGULAIR ist bei Patienten mit Überempfindlichkeit gegen einen seiner Bestandteile kontraindiziert.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Die Cysteinyl-Leukotriene (LTC4, LTD4, LTE4) sind Produkte des Arachidonsäure-Stoffwechsels und werden von verschiedenen Zellen, einschließlich Mastzellen und Eosinophilen, freigesetzt. Diese Eicosanoide binden an Cysteinyl-Leukotrien (CysLT)-Rezeptoren. Der CysLT-Typ-1 (CysLT1)-Rezeptor findet sich in den menschlichen Atemwegen (einschließlich glatter Atemwegsmuskelzellen und Atemwegsmakrophagen) und auf anderen entzündungsfördernden Zellen (einschließlich Eosinophilen und bestimmten myeloischen Stammzellen). CysLTs wurden mit der Pathophysiologie von Asthma und allergischer Rhinitis korreliert. Bei Asthma umfassen Leukotrien-vermittelte Wirkungen Atemwegsödeme, Kontraktion der glatten Muskulatur und veränderte Zellaktivität im Zusammenhang mit dem Entzündungsprozess. Bei allergischer Rhinitis werden CysLTs nach Allergenexposition sowohl während der Früh- als auch der Spätphasenreaktion aus der Nasenschleimhaut freigesetzt und sind mit Symptomen einer allergischen Rhinitis verbunden.
Montelukast ist eine oral wirksame Verbindung, die mit hoher Affinität und Selektivität an den CysLT1-Rezeptor bindet (vorzugsweise gegenüber anderen pharmakologisch wichtigen Atemwegsrezeptoren wie dem Prostanoid-, cholinergen oder β-adrenergen Rezeptor). Montelukast hemmt die physiologischen Wirkungen von LTD4 am CysLT1-Rezeptor ohne agonistische Aktivität.
Pharmakodynamik
Montelukast bewirkt eine Hemmung der Cysteinyl-Leukotrien-Rezeptoren der Atemwege, wie durch die Fähigkeit zur Hemmung der Bronchokonstriktion aufgrund von inhaliertem LTD4 bei Asthmatikern gezeigt wurde. So niedrige Dosen wie 5 mg verursachen eine erhebliche Blockade der LTD4-induzierten Bronchokonstriktion. In einer Placebo-kontrollierten Crossover-Studie (n=12) hemmte SINGULAIR 4 mg die Bronchokonstriktion in der Früh- und Spätphase aufgrund einer Antigenprovokation um 75 % bzw. 57 %.
Die Wirkung von SINGULAIR 5 mg auf Eosinophile im peripheren Blut wurde in klinischen Studien untersucht. Bei Patienten mit Asthma ab 2 Jahren, die SINGULAIR 10 mg erhielten, wurde im Vergleich zu Placebo während der doppelblinden Behandlungsperioden eine Abnahme der mittleren Eosinophilenzahl im peripheren Blut um 9 % bis 15 % festgestellt. Bei Patienten mit saisonaler allergischer Rhinitis im Alter von 15 Jahren und älter, die SINGULAIR 4 mg erhielten, wurde im Vergleich zur doppelblinden Behandlung eine mittlere Zunahme der Eosinophilenzahl im peripheren Blut um 0,2 % festgestellt, verglichen mit einer mittleren Zunahme von 12,5 % bei den mit Placebo behandelten Patienten Perioden; dies entspricht einer mittleren Differenz von 12,3 % zugunsten von SINGULAIR. Der Zusammenhang zwischen diesen Beobachtungen und den in den klinischen Studien festgestellten klinischen Vorteilen von Montelukast ist nicht bekannt [siehe Klinische Studien ].
Pharmakokinetik
Absorption
Montelukast wird nach oraler Verabreichung schnell resorbiert. Nach Verabreichung der 10-mg-Filmtablette an nüchternen Erwachsenen wird die mittlere maximale Montelukast-Plasmakonzentration (Cmax) innerhalb von 3 bis 4 Stunden (Tmax) erreicht. Die mittlere orale Bioverfügbarkeit beträgt 64 %. Die orale Bioverfügbarkeit und Cmax werden durch eine Standardmahlzeit am Morgen nicht beeinflusst.
Bei der 5-mg-Kautablette wird die mittlere Cmax 2 bis 2,5 Stunden nach der Verabreichung bei Erwachsenen im nüchternen Zustand erreicht. Die mittlere orale Bioverfügbarkeit beträgt 73 % im nüchternen Zustand gegenüber 63 % bei Verabreichung mit einer Standardmahlzeit am Morgen.
Für die 4-mg-Kautablette wird die mittlere Cmax 2 Stunden nach der Verabreichung bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren im nüchternen Zustand erreicht.
Die orale 4-mg-Granulatformulierung ist bioäquivalent zu der 4-mg-Kautablette, wenn sie Erwachsenen im nüchternen Zustand verabreicht wird. Die gleichzeitige Verabreichung der oralen Granulatformulierung mit Apfelmus hatte keine klinisch signifikante Wirkung auf die Pharmakokinetik von Montelukast. Eine fettreiche Mahlzeit am Morgen hatte keinen Einfluss auf die AUC von Montelukast Granulat zum Einnehmen; Die Mahlzeit verringerte jedoch Cmax um 35 % und verlängerte Tmax von 2,3 ± 1,0 Stunden auf 6,4 ± 2,9 Stunden.
Die Sicherheit und Wirksamkeit von SINGULAIR bei Patienten mit Asthma wurde in klinischen Studien nachgewiesen, in denen die 10-mg-Filmtabletten und die 5-mg-Kautabletten-Formulierungen abends unabhängig vom Zeitpunkt der Nahrungsaufnahme verabreicht wurden. Die Sicherheit von SINGULAIR 4 mg bei Patienten mit Asthma wurde auch in klinischen Studien nachgewiesen, in denen die 4-mg-Kautablette und die orale 4-mg-Granulatformulierung abends unabhängig vom Zeitpunkt der Nahrungsaufnahme verabreicht wurden. Die Sicherheit und Wirksamkeit von SINGULAIR bei Patienten mit saisonaler allergischer Rhinitis wurden in klinischen Studien nachgewiesen, in denen die 10-mg-Filmtablette morgens oder abends unabhängig vom Zeitpunkt der Nahrungsaufnahme eingenommen wurde.
Die vergleichende Pharmakokinetik von Montelukast bei Verabreichung als zwei 5-mg-Kautabletten gegenüber einer 10-mg-Filmtablette wurde nicht untersucht.
Verteilung
Montelukast wird zu mehr als 99 % an Plasmaproteine gebunden. Das Verteilungsvolumen von Montelukast im Steady State beträgt durchschnittlich 8 bis 11 Liter. Oral verabreichtes Montelukast verteilt sich bei Ratten im Gehirn.
Beseitigung
Die Plasma-Clearance von Montelukast beträgt bei gesunden Erwachsenen durchschnittlich 45 ml/min. Nach einer oralen Dosis von radioaktiv markiertem Montelukast wurden 86 % der Radioaktivität in 5-tägigen Stuhlsammlungen und
In mehreren Studien lag die mittlere Plasmahalbwertszeit von Montelukast bei gesunden jungen Erwachsenen zwischen 2,7 und 5,5 Stunden. Die Pharmakokinetik von Montelukast ist bei oralen Dosen bis zu 50 mg nahezu linear. Bei einmal täglicher Gabe von 10 mg Montelukast kommt es zu einer geringen Akkumulation der Muttersubstanz im Plasma (14 %).
Stoffwechsel
Montelukast wird extensiv metabolisiert. In Studien mit therapeutischen Dosen waren Plasmakonzentrationen von Montelukast-Metaboliten bei Erwachsenen und pädiatrischen Patienten im Steady State nicht nachweisbar.
In-vitro-Studien mit menschlichen Lebermikrosomen weisen darauf hin, dass CYP3A4, 2C8 und 2C9 am Metabolismus von Montelukast beteiligt sind. In klinisch relevanten Konzentrationen scheint 2C8 eine wichtige Rolle im Metabolismus von Montelukast zu spielen.
Spezifische Populationen
Patienten mit eingeschränkter Leberfunktion
Patienten mit leichter bis mittelschwerer Leberinsuffizienz und klinischem Nachweis einer Zirrhose zeigten Hinweise auf eine verminderte Metabolisierung von Montelukast, was zu einer um 41 % (90 % KI = 7 %, 85 %) höheren mittleren AUC von Montelukast nach einer Einzeldosis von 10 mg führte. Die Elimination von Montelukast war im Vergleich zu gesunden Probanden leicht verlängert (mittlere Halbwertszeit 7,4 Stunden). Bei Patienten mit leichter bis mittelschwerer Leberinsuffizienz ist keine Dosisanpassung erforderlich. Die Pharmakokinetik von SINGULAIR bei Patienten mit schwerer Leberfunktionsstörung oder Hepatitis wurde nicht untersucht.
Patienten mit eingeschränkter Nierenfunktion
Da Montelukast und seine Metaboliten nicht im Urin ausgeschieden werden, wurde die Pharmakokinetik von Montelukast bei Patienten mit Niereninsuffizienz nicht untersucht. Bei diesen Patienten wird keine Dosisanpassung empfohlen.
Männliche und weibliche Patienten
Die Pharmakokinetik von Montelukast ist bei Männern und Frauen ähnlich.
Rassengruppen
Unterschiede in der Pharmakokinetik aufgrund der Rasse wurden nicht untersucht.
Jugendliche und pädiatrische Patienten
Pharmakokinetische Studien untersuchten die systemische Exposition der oralen 4-mg-Granulatformulierung bei pädiatrischen Patienten im Alter von 6 bis 23 Monaten, der 4-mg-Kautabletten bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren und der 5-mg-Kautabletten bei pädiatrischen Patienten 6 bis 14 Jahre und die 10-mg-Filmtabletten bei jungen Erwachsenen und Jugendlichen ≥ 15 Jahren.
Das Plasmakonzentrationsprofil von Montelukast nach Verabreichung der 10-mg-Filmtablette ist bei Jugendlichen ≥ 15 Jahren und jungen Erwachsenen ähnlich. Die 10-mg-Filmtablette wird zur Anwendung bei Patienten im Alter von ≥ 15 Jahren empfohlen.
Die mittlere systemische Exposition der 4-mg-Kautablette bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren und der 5-mg-Kautablette bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren ist ähnlich der mittleren systemischen Exposition der 10-mg-Filmtablette. überzogene Tablette bei Erwachsenen. Die 5-mg-Kautablette sollte bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren und die 4-mg-Kautablette sollte bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren angewendet werden.
Bei Kindern im Alter von 6 bis 11 Monaten waren die systemische Montelukast-Exposition und die Variabilität der Montelukast-Plasmakonzentrationen höher als bei Erwachsenen. Basierend auf Populationsanalysen war die mittlere AUC (4296 ng•h/ml [Bereich 1200 bis 7153]) um 60 % höher und die mittlere Cmax (667 ng/ml [Bereich 201 bis 1058]) um 89 % höher als die in beobachteten Erwachsene (mittlere AUC 2689 ng•h/ml [Bereich 1521 bis 4595]) und mittlere Cmax (353 ng/ml [Bereich 180 bis 548]). Die systemische Exposition bei Kindern im Alter von 12 bis 23 Monaten war weniger variabel, aber immer noch höher als bei Erwachsenen. Die mittlere AUC (3574 ng•h/ml [Bereich 2229 bis 5408]) war 33 % höher und die mittlere Cmax (562 ng/ml [Bereich 296 bis 814]) war 60 % höher als bei Erwachsenen. Sicherheit und Verträglichkeit von Montelukast in einer pharmakokinetischen Studie mit Einzeldosis bei 26 Kindern im Alter von 6 bis 23 Monaten waren ähnlich wie bei Patienten ab zwei Jahren [siehe NEBENWIRKUNGEN ]. Die orale 4-mg-Granulatformulierung sollte bei pädiatrischen Patienten im Alter von 12 bis 23 Monaten zur Behandlung von Asthma oder bei pädiatrischen Patienten im Alter von 6 bis 23 Monaten zur Behandlung von ganzjähriger allergischer Rhinitis angewendet werden. Da die orale 4-mg-Granulatformulierung mit der 4-mg-Kautablette bioäquivalent ist, kann sie bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren auch als alternative Formulierung zur 4-mg-Kautablette verwendet werden.
Arzneimittelwechselwirkungsstudien
Theophyllin, Prednison und Prednisolon
SINGULAIR wurde zusammen mit anderen Therapien verabreicht, die routinemäßig zur Prophylaxe und chronischen Behandlung von Asthma eingesetzt werden, ohne erkennbare Zunahme von Nebenwirkungen. In Wechselwirkungsstudien hatte die empfohlene klinische Dosis von Montelukast keine klinisch bedeutsamen Auswirkungen auf die Pharmakokinetik der folgenden Arzneimittel: Theophyllin, Prednison und Prednisolon.
Montelukast in einer Dosis von 10 mg einmal täglich bis zum pharmakokinetischen Steady State verursachte keine klinisch signifikanten Veränderungen der Kinetik einer intravenösen Einzeldosis von Theophyllin [vorwiegend ein Cytochrom P450 (CYP) 1A2-Substrat]. Montelukast in Dosen von ≥ 100 mg täglich bis zum pharmakokinetischen Steady State verursachte keine klinisch signifikante Veränderung der Plasmaprofile von Prednison oder Prednisolon nach Verabreichung von entweder oralem Prednison oder intravenösem Prednisolon.
Orale Kontrazeptiva, Fexofenadin, Digoxin und Warfarin
In Wechselwirkungsstudien hatte die empfohlene klinische Dosis von Montelukast keine klinisch relevanten Auswirkungen auf die Pharmakokinetik der folgenden Arzneimittel: orale Kontrazeptiva (Norethindron 1 mg/Ethinylestradiol 35 mcg), Digoxin und Warfarin. Montelukast in Dosen von ≥ 100 mg täglich bis zum pharmakokinetischen Steady State veränderte die Plasmakonzentrationen der beiden Komponenten eines oralen Kontrazeptivums, das Norethindron 1 mg/Ethinylestradiol 35 µg enthielt, nicht signifikant. Montelukast in einer Dosis von 10 mg einmal täglich bis zum pharmakokinetischen Steady State veränderte das Plasmakonzentrationsprofil von Fexofenadin, das pharmakokinetische Profil oder die Urinausscheidung von immunreaktivem Digoxin nicht; veränderte weder das pharmakokinetische Profil von Warfarin (hauptsächlich ein Substrat von CYP2C9, 3A4 und 1A2) noch beeinflusste es die Wirkung einer oralen Einzeldosis von 30 mg Warfarin auf die Prothrombinzeit oder die International Normalized Ratio (INR).
Schilddrüsenhormone, beruhigende Hypnotika, nichtsteroidale Entzündungshemmer, Benzodiazepine und abschwellende Mittel
Obwohl keine zusätzlichen spezifischen Wechselwirkungsstudien durchgeführt wurden, wurde SINGULAIR in klinischen Studien gleichzeitig mit einer Vielzahl häufig verschriebener Arzneimittel angewendet, ohne dass es Hinweise auf unerwünschte klinische Wechselwirkungen gab. Zu diesen Medikamenten gehörten Schilddrüsenhormone, sedierende Hypnotika, nichtsteroidale entzündungshemmende Mittel, Benzodiazepine und abschwellende Mittel.
Cytochrom P450 (CYP) Enzyminduktoren
Phenobarbital, das den Leberstoffwechsel induziert, verringerte die Fläche unter der Plasmakonzentrationskurve (AUC) von Montelukast nach einer Einzeldosis von 10 mg Montelukast um etwa 40 %. Es wird keine Dosisanpassung für SINGULAIR empfohlen. Es ist sinnvoll, eine angemessene klinische Überwachung durchzuführen, wenn starke CYP-Enzyminduktoren wie Phenobarbital oder Rifampin zusammen mit SINGULAIR verabreicht werden.
Wirkung von Montelukast auf Cytochrom P450 (CYP) Enzyme
Montelukast ist in vitro ein starker Inhibitor von CYP2C8. Daten aus einer klinischen Wechselwirkungsstudie mit Montelukast und Rosiglitazon (ein Sondensubstrat, das repräsentativ für hauptsächlich durch CYP2C8 metabolisierte Arzneimittel ist) bei 12 gesunden Personen zeigten jedoch, dass die Pharmakokinetik von Rosiglitazon bei gleichzeitiger Verabreichung der Arzneimittel nicht verändert wird, was darauf hindeutet, dass Montelukast dies tut CYP2C8 in vivo nicht hemmen. Daher ist nicht zu erwarten, dass Montelukast den Metabolismus von Arzneimitteln verändert, die durch dieses Enzym metabolisiert werden (z. B. Paclitaxel, Rosiglitazon und Repaglinid). Basierend auf weiteren In-vitro-Ergebnissen in menschlichen Lebermikrosomen hemmen therapeutische Plasmakonzentrationen von Montelukast CYP 3A4, 2C9, 1A2, 2A6, 2C19 oder 2D6 nicht.
Cytochrom P450 (CYP) Enzyminhibitoren
In-vitro-Studien haben gezeigt, dass Montelukast ein Substrat von CYP 2C8, 2C9 und 3A4 ist. Die gleichzeitige Anwendung von Montelukast mit Itraconazol, einem starken CYP 3A4-Hemmer, führte zu keiner signifikanten Erhöhung der systemischen Exposition von Montelukast. Daten aus einer klinischen Wechselwirkungsstudie mit Montelukast und Gemfibrozil (einem Inhibitor von CYP 2C8 und 2C9) zeigten, dass Gemfibrozil in therapeutischer Dosis die systemische Exposition von Montelukast um das 4,4-Fache erhöhte. Die gleichzeitige Anwendung von Itraconazol, Gemfibrozil und Montelukast erhöhte die systemische Exposition von Montelukast nicht weiter. Basierend auf den verfügbaren klinischen Erfahrungen ist keine Dosisanpassung von Montelukast bei gleichzeitiger Anwendung mit Gemfibrozil erforderlich [siehe ÜBERDOSIS ].
Klinische Studien
Asthma
Erwachsene und Jugendliche ab 15 Jahren mit Asthma
Klinische Studien mit Erwachsenen und Jugendlichen ab 15 Jahren haben gezeigt, dass Montelukast-Dosen über 10 mg einmal täglich keinen zusätzlichen klinischen Nutzen bringen.
Die Wirksamkeit von SINGULAIR zur chronischen Behandlung von Asthma bei Erwachsenen und Jugendlichen ab 15 Jahren wurde in zwei (USA und multinationalen) ähnlich angelegten, randomisierten, 12-wöchigen, doppelblinden, placebokontrollierten Studien mit 1576 Patienten nachgewiesen ( 795 mit SINGULAIR behandelt, 530 mit Placebo behandelt und 251 mit aktiver Kontrolle behandelt). Das Durchschnittsalter betrug 33 Jahre (Bereich 15 bis 85); 56,8 % waren weiblich und 43,2 % männlich. Die ethnische/rassische Verteilung in diesen Studien war 71,6 % Kaukasier, 17,7 % Hispanoamerikaner, 7,2 % anderer Herkunft und 3,5 % Schwarze. Die Patienten hatten leichtes oder mittelschweres Asthma und waren Nichtraucher, die nach Bedarf etwa 5 Sprühstöße des inhalierten β-Agonisten pro Tag benötigten. Die Patienten hatten einen mittleren prozentualen Baseline-Prozentsatz des vorhergesagten forcierten Exspirationsvolumens in 1 Sekunde (FEV1) von 66 % (ungefährer Bereich 40 bis 90 %). Die co-primären Endpunkte in diesen Studien waren FEV1 und Asthmasymptome am Tag. In beiden Studien wurde nach 12 Wochen eine zufällig ausgewählte Untergruppe von Patienten, die SINGULAIR 5 mg erhielten, für weitere 3 Wochen einer doppelblinden Behandlung auf Placebo umgestellt, um mögliche Rebound-Effekte zu untersuchen.
Die Ergebnisse der US-Studie zum primären Endpunkt, dem morgendlichen FEV1, ausgedrückt als mittlere prozentuale Veränderung gegenüber dem Ausgangswert, gemittelt über den 12-wöchigen Behandlungszeitraum, sind in ABBILDUNG 2 dargestellt. Im Vergleich zu Placebo wurde die Behandlung mit einer SINGULAIR 10-mg-Tablette täglich in am Abend führte zu einem statistisch signifikanten Anstieg der FEV1-Veränderung in Prozent gegenüber dem Ausgangswert (13,0 %-Veränderung in der mit SINGULAIR 4 mg behandelten Gruppe vs. 4,2 %-Veränderung in der Placebogruppe, p
Abbildung 2: Mittlere prozentuale Veränderung des FEV1 gegenüber dem Ausgangswert (US-Studie: SINGULAIR 5 mg N = 406; Placebo N = 270) (ANOVA-Modell)
Die Wirkung von SINGULAIR 5 mg auf andere primäre und sekundäre Endpunkte der multinationalen Studie ist in TABELLE 6 dargestellt. Die Ergebnisse zu diesen Endpunkten waren in der US-Studie ähnlich.
Beide Studien bewerteten die Wirkung von SINGULAIR 4 mg auf sekundäre Ergebnisse, einschließlich Asthmaanfall (Inanspruchnahme von Gesundheitsversorgungsressourcen wie ein außerplanmäßiger Besuch in einer Arztpraxis, Notaufnahme oder einem Krankenhaus; oder Behandlung mit oralen, intravenösen oder intramuskuläres Kortikosteroid) und die Anwendung von oralen Kortikosteroiden zur Asthmarettung. In der multinationalen Studie erlitten signifikant weniger Patienten (15,6 % der Patienten) unter SINGULAIR Asthmaanfälle im Vergleich zu Patienten unter Placebo (27,3 %, p
Wirkungseintritt und Aufrechterhaltung der Wirkung
In jeder Placebo-kontrollierten Studie mit Erwachsenen wurde der Behandlungseffekt von SINGULAIR 4 mg, gemessen anhand der Parameter der täglichen Tagebuchkarte, einschließlich Symptom-Scores, bedarfsgerechter Anwendung von β-Agonisten und PEFR-Messungen, nach der ersten Dosis erreicht und hielt an während des gesamten Dosierungsintervalls (24 Stunden). In nicht placebokontrollierten Verlängerungsstudien über einen Zeitraum von bis zu einem Jahr wurde während der kontinuierlichen einmal täglichen abendlichen Verabreichung keine signifikante Veränderung des Behandlungseffekts beobachtet. Das Absetzen von SINGULAIR 4 mg bei Asthmapatienten nach 12-wöchiger kontinuierlicher Anwendung führte nicht zu einer Rebound-Verschlechterung des Asthmas.
Pädiatrische Patienten im Alter von 6 bis 14 Jahren mit Asthma
Die Wirksamkeit von SINGULAIR 5 mg bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren wurde in einer 8-wöchigen, doppelblinden, placebokontrollierten Studie mit 336 Patienten (201 behandelt mit SINGULAIR und 135 behandelt mit Placebo) unter Verwendung eines inhalativen β-Agonisten nachgewiesen auf einer „nach Bedarf“-Basis. Die Patienten hatten einen mittleren prozentualen FEV1-Ausgangswert von 72 % (ungefährer Bereich 45 bis 90 %) und einen mittleren täglichen Bedarf an inhalativen β-Agonisten von 3,4 Sprühstößen Albuterol. Etwa 36 % der Patienten erhielten inhalative Kortikosteroide. Das Durchschnittsalter betrug 11 Jahre (Bereich 6 bis 15); 35,4 % waren weiblich und 64,6 % männlich. Die ethnische/rassische Verteilung in dieser Studie war 80,1 % Kaukasier, 12,8 % Schwarze, 4,5 % Hispanoamerikaner und 2,7 % anderer Herkunft.
Im Vergleich zu Placebo führte die Behandlung mit einer 5-mg-Kautablette SINGULAIR 4 mg täglich zu einer signifikanten Verbesserung der prozentualen Veränderung des mittleren morgendlichen FEV1 gegenüber dem Ausgangswert (8,7 % in der mit SINGULAIR 4 mg behandelten Gruppe gegenüber 4,2 % Veränderung gegenüber dem Ausgangswert in der Placebo-Gruppe). p
Ähnlich wie in den Studien mit Erwachsenen wurde in einer unverblindeten Verlängerungsstudie ohne gleichzeitige Placebogruppe über bis zu 6 Monate keine signifikante Veränderung des Behandlungseffekts bei kontinuierlicher einmal täglicher Verabreichung beobachtet.
Pädiatrische Patienten im Alter von 2 bis 5 Jahren mit Asthma
Die Wirksamkeit von SINGULAIR zur chronischen Behandlung von Asthma bei pädiatrischen Patienten im Alter von 2 bis 5 Jahren wurde in einer 12-wöchigen placebokontrollierten Sicherheits- und Verträglichkeitsstudie mit 689 Patienten untersucht, von denen 461 mit SINGULAIR behandelt wurden. Das Durchschnittsalter betrug 4 Jahre (Bereich 2 bis 6); 41,5 % waren weiblich und 58,5 % männlich. Die ethnische/rassische Verteilung in dieser Studie war 56,5 % Kaukasier, 20,9 % Hispanoamerikaner, 14,4 % anderer Herkunft und 8,3 % Schwarze.
Während das primäre Ziel darin bestand, die Sicherheit und Verträglichkeit von SINGULAIR 10 mg in dieser Altersgruppe zu bestimmen, umfasste die Studie explorative Wirksamkeitsbewertungen, einschließlich Asthmasymptom-Scores am Tag und über Nacht, Anwendung von β-Agonisten, orale Kortikosteroid-Rescue und eine umfassende Bewertung durch den Arzt. Die Ergebnisse dieser explorativen Wirksamkeitsbewertungen zusammen mit der Pharmakokinetik und Extrapolation von Wirksamkeitsdaten älterer Patienten stützen die Gesamtschlussfolgerung, dass SINGULAIR bei der Erhaltungsbehandlung von Asthma bei Patienten im Alter von 2 bis 5 Jahren wirksam ist.
Wirkungen bei Patienten mit gleichzeitiger inhalativer Kortikosteroide
In separaten Studien an Erwachsenen wurde die Fähigkeit von SINGULAIR 10 mg untersucht, die klinische Wirkung von inhalativen Kortikosteroiden zu verstärken und das Ausschleichen der inhalativen Kortikosteroide bei gleichzeitiger Anwendung zu ermöglichen.
In eine randomisierte, placebokontrollierte Parallelgruppenstudie (n = 226) wurden Erwachsene mit stabilem Asthma mit einem mittleren FEV1 von etwa 84 % des Sollwerts aufgenommen, die zuvor verschiedene inhalative Kortikosteroide (verabreicht durch Dosieraerosole oder Trockenpulverinhalatoren) erhalten hatten ). Das Durchschnittsalter betrug 41,5 Jahre (Bereich 16 bis 70); 52,2 % waren weiblich und 47,8 % männlich. Die ethnische/rassische Verteilung in dieser Studie war 92,0 % Kaukasier, 3,5 % Schwarze, 2,2 % Hispanoamerikaner und 2,2 % Asiaten. Die Arten von inhalativen Kortikosteroiden und ihr mittlerer Ausgangsbedarf umfassten Beclomethasondipropionat (mittlere Dosis 1203 Mikrogramm/Tag), Triamcinolonacetonid (mittlere Dosis 2004 Mikrogramm/Tag), Flunisolid (mittlere Dosis 1971 Mikrogramm/Tag), Fluticasonpropionat (mittlere Dosis 1971 Mikrogramm/Tag). Dosis 1083 µg/Tag) oder Budesonid (mittlere Dosis 1192 µg/Tag). Bei einigen dieser inhalativen Kortikosteroide handelte es sich um nicht in den USA zugelassene Formulierungen, und die angegebenen Dosen sind möglicherweise nicht ex-actuator. Der Bedarf an inhalativen Kortikosteroiden vor der Studie wurde während einer 5- bis 7-wöchigen Placebo-Run-in-Phase um etwa 37 % reduziert, um die Patienten auf ihre niedrigste wirksame inhalative Kortikosteroiddosis zu titrieren. Die Behandlung mit SINGULAIR 4 mg führte zu einer weiteren Reduktion der mittleren inhalativen Kortikosteroiddosis um 47 % im Vergleich zu einer mittleren Reduktion von 30 % in der Placebogruppe über den 12-wöchigen aktiven Behandlungszeitraum (p ≤ 0,05). Es ist nicht bekannt, ob die Ergebnisse dieser Studie auf Patienten mit Asthma verallgemeinert werden können, die höhere Dosen von inhalativen Kortikosteroiden oder systemischen Kortikosteroiden benötigen.
In einer anderen randomisierten, placebokontrollierten Parallelgruppenstudie (n = 642) in einer ähnlichen Population von erwachsenen Patienten, die zuvor mit inhalativen Kortikosteroiden (Beclomethason 336 µg/Tag) behandelt, aber nicht angemessen kontrolliert wurden, wurde SINGULAIR zusätzlich zu Beclomethason verabreicht in statistisch signifikanten Verbesserungen des FEV1 im Vergleich zu den Patienten, die Beclomethason allein weiter erhielten, oder den Patienten, die Beclomethason abgesetzt und in den letzten 10 Wochen der 16-wöchigen, verblindeten Behandlungsphase nur mit Montelukast oder Placebo behandelt wurden. Patienten, die randomisiert den Behandlungsarmen mit Beclomethason zugeteilt wurden, hatten eine statistisch signifikant bessere Asthmakontrolle als die Patienten, die randomisiert SINGULAIR allein oder Placebo allein erhielten, wie durch FEV1, Asthmasymptome am Tag, PEFR, nächtliches Erwachen aufgrund von Asthma und „nach Bedarf“-β- angezeigt. agonistische Anforderungen.
Bei erwachsenen Patienten mit Asthma mit dokumentierter Aspirin-Empfindlichkeit, die fast alle gleichzeitig inhalative und/oder orale Kortikosteroide erhielten, zeigte eine 4-wöchige, randomisierte Parallelgruppenstudie (n=80), dass SINGULAIR 10 mg im Vergleich zu Placebo zu Ergebnissen führte zu einer signifikanten Verbesserung der Parameter der Asthmakontrolle. Das Ausmaß der Wirkung von SINGULAIR 4 mg bei Aspirin-empfindlichen Patienten war ähnlich der Wirkung, die bei der allgemeinen untersuchten Population von Asthmapatienten beobachtet wurde. Die Wirkung von SINGULAIR auf die bronchokonstriktorische Reaktion auf Aspirin oder andere nicht-steroidale entzündungshemmende Arzneimittel bei Aspirin-empfindlichen Asthmapatienten wurde nicht untersucht [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Belastungsinduzierte Bronchokonstriktion (EIB)
Belastungsinduzierte Bronchokonstriktion (Erwachsene, Jugendliche und pädiatrische Patienten ab 6 Jahren)
Die Wirksamkeit von SINGULAIR 5 mg, 10 mg bei Verabreichung als Einzeldosis 2 Stunden vor dem Training zur Vorbeugung von EIB wurde in drei (USA und multinationalen), randomisierten, doppelblinden, Placebo-kontrollierten Crossover-Studien mit insgesamt untersucht 160 erwachsene und jugendliche Patienten ab 15 Jahren mit EIB. Belastungstests wurden 2 Stunden, 8,5 oder 12 Stunden und 24 Stunden nach Verabreichung einer Einzeldosis des Studienmedikaments (SINGULAIR 10 mg oder Placebo) durchgeführt. Der primäre Endpunkt war in allen drei Studien (Studie A, Studie B und Studie C) der mittlere maximale prozentuale Abfall des FEV1 nach der Belastungsbelastung 2 Stunden nach der Dosisgabe. In Studie A zeigte eine Einzeldosis von SINGULAIR 10 mg einen statistisch signifikanten Schutzvorteil gegen EIB, wenn sie 2 Stunden vor dem Training eingenommen wurde. Einige Patienten waren 8,5 und 24 Stunden nach der Verabreichung vor EIB geschützt; Einige Patienten waren es jedoch nicht. Die Ergebnisse für den mittleren maximalen prozentualen Abfall zu jedem Zeitpunkt in Studie A sind in TABELLE 7 gezeigt und sind repräsentativ für die Ergebnisse der anderen beiden Studien.
Die Wirksamkeit von SINGULAIR 5-mg-Kautabletten bei Verabreichung als Einzeldosis 2 Stunden vor dem Training zur Vorbeugung von EIB wurde in einer multinationalen, randomisierten, doppelblinden, placebokontrollierten Crossover-Studie mit insgesamt 64 pädiatrischen Patienten untersucht Patienten im Alter von 6 bis 14 Jahren mit EIB. Belastungsbelastungstests wurden 2 Stunden und 24 Stunden nach Verabreichung einer Einzeldosis des Studienmedikaments (SINGULAIR 5 mg oder Placebo) durchgeführt. Der primäre Endpunkt war der mittlere maximale prozentuale Abfall des FEV1 nach der Belastungsbelastung 2 Stunden nach der Dosisgabe. Eine Einzeldosis von SINGULAIR 5 mg zeigte einen statistisch signifikanten Schutzvorteil gegen EIB, wenn sie 2 Stunden vor dem Training eingenommen wurde (TABELLE 8). Ähnliche Ergebnisse wurden 24 Stunden nach der Einnahme gezeigt (ein sekundärer Endpunkt). Einige Patienten waren 24 Stunden nach der Verabreichung vor EIB geschützt; Einige Patienten waren es jedoch nicht. Es wurden keine Zeitpunkte zwischen 2 und 24 Stunden nach der Verabreichung bewertet.
Die Wirksamkeit von SINGULAIR 5 mg zur Vorbeugung von EIB bei Patienten unter 6 Jahren wurde nicht nachgewiesen.
Die tägliche Verabreichung von SINGULAIR 4 mg zur chronischen Behandlung von Asthma ist nicht dafür geeignet, akute Episoden von EIB zu verhindern.
In einer 12-wöchigen, randomisierten, doppelblinden Parallelgruppenstudie mit 110 erwachsenen und jugendlichen Asthmatikern im Alter von 15 Jahren und älter mit einem mittleren FEV1-Prozentsatz von 83 % zu Studienbeginn und mit dokumentierter belastungsinduzierter Exazerbation von Asthma mit SINGULAIR, 10 mg, einmal täglich am Abend, führte zu einer statistisch signifikanten Verringerung des mittleren maximalen prozentualen Abfalls des FEV1 und der mittleren Zeit bis zur Erholung auf innerhalb von 5 % des FEV1 vor dem Training. Am Ende des Dosierungsintervalls (dh 20 bis 24 Stunden nach der vorhergehenden Dosis) wurde eine Belastungsbelastung durchgeführt. Diese Wirkung hielt während des 12-wöchigen Behandlungszeitraums an, was darauf hindeutet, dass keine Toleranz auftrat. SINGULAIR 4 mg verhinderte jedoch bei 52 % der untersuchten Patienten keine klinisch signifikante Verschlechterung des maximalen prozentualen Abfalls des FEV1 nach Belastung (dh ≥ 20 % Abnahme gegenüber dem Ausgangswert vor der Belastung). In einer separaten Crossover-Studie bei Erwachsenen wurde eine ähnliche Wirkung nach zweimal täglicher Gabe von 10 mg SINGULAIR beobachtet.
Bei pädiatrischen Patienten im Alter von 6 bis 14 Jahren, die die 5-mg-Kautablette einnahmen, zeigte eine 2-tägige Crossover-Studie ähnliche Wirkungen wie bei Erwachsenen, wenn am Ende des Dosierungsintervalls (d. h. 20 bis 24 Stunden nach der vorherigen Dosis).
Allergische Rhinitis (saisonal und mehrjährig)
Saisonale allergische Rhinitis
Die Wirksamkeit von SINGULAIR Tabletten zur Behandlung von saisonaler allergischer Rhinitis wurde in 5 ähnlich angelegten, randomisierten, doppelblinden, placebo- und wirkstoffkontrollierten (Loratadin) Parallelgruppenstudien untersucht, die in Nordamerika durchgeführt wurden. In die 5 Studien wurden insgesamt 5029 Patienten aufgenommen, von denen 1799 mit SINGULAIR-Tabletten behandelt wurden. Die Patienten waren 15 bis 82 Jahre alt und hatten bei Studienbeginn eine Vorgeschichte mit saisonaler allergischer Rhinitis, einen positiven Hauttest auf mindestens ein relevantes saisonales Allergen und aktive Symptome einer saisonalen allergischen Rhinitis.
Die Dauer der randomisierten Behandlung betrug 2 Wochen in 4 Studien und 4 Wochen in einer Studie. Die primäre Ergebnisvariable war die durchschnittliche Veränderung gegenüber dem Ausgangswert des Scores für nasale Symptome am Tag (der Durchschnitt der individuellen Scores für verstopfte Nase, Rhinorrhö, Nasenjucken, Niesen), die von den Patienten auf einer kategorialen Skala von 0 bis 3 bewertet wurden.
Vier der fünf Studien zeigten mit SINGULAIR 10-mg-Tabletten im Vergleich zu Placebo eine signifikante Verringerung der Scores für nasale Symptome am Tag. Die Ergebnisse eines Versuchs sind unten gezeigt. Das Durchschnittsalter in dieser Studie betrug 35,0 Jahre (Bereich 15 bis 81); 65,4 % waren weiblich und 34,6 % männlich. Die ethnische/rassische Verteilung in dieser Studie war 83,1 % Kaukasier, 6,4 % anderer Herkunft, 5,8 % Schwarze und 4,8 % Hispanoamerikaner. Die durchschnittlichen Veränderungen gegenüber dem Ausgangswert des Scores für nasale Symptome am Tag in den Behandlungsgruppen, die SINGULAIR 4 mg Tabletten, Loratadin und Placebo erhielten, sind in TABELLE 9 dargestellt. Die verbleibenden drei Studien, die die Wirksamkeit zeigten, zeigten ähnliche Ergebnisse. Die Wirksamkeit wurde bei saisonaler allergischer Rhinitis nachgewiesen, wenn Montelukast morgens oder abends verabreicht wurde.
Ganzjährige allergische Rhinitis
Die Wirksamkeit von SINGULAIR 5 mg Tabletten zur Behandlung von ganzjähriger allergischer Rhinitis wurde in zwei randomisierten, doppelblinden, placebokontrollierten Studien untersucht, die in Nordamerika und Europa durchgeführt wurden. In die beiden Studien wurden insgesamt 3357 Patienten aufgenommen, von denen 1632 SINGULAIR 10-mg-Tabletten erhielten. Patienten im Alter von 15 bis 82 Jahren mit ganzjähriger allergischer Rhinitis, bestätigt durch die Anamnese und einen positiven Hauttest auf mindestens ein relevantes ganzjähriges Allergen (Hausstaubmilben, Tierhaare und/oder Schimmelpilzsporen), die zum Zeitpunkt der Studie aktive Symptome hatten Eintrag, wurden eingeschrieben.
In der Studie, in der die Wirksamkeit nachgewiesen wurde, betrug das mediane Alter 35 Jahre (Bereich 15 bis 81); 64,1 % waren weiblich und 35,9 % männlich. Die ethnische/rassische Verteilung in dieser Studie war 83,2 % Kaukasier, 8,1 % Schwarze, 5,4 % Hispanoamerikaner, 2,3 % Asiaten und 1,0 % anderer Herkunft. Es wurde gezeigt, dass SINGULAIR 10 mg Tabletten einmal täglich die Symptome der ganzjährigen allergischen Rhinitis über einen Behandlungszeitraum von 6 Wochen signifikant reduziert (TABELLE 10); In dieser Studie war die primäre Ergebnisvariable die mittlere Veränderung des Scores für nasale Symptome am Tag (der Durchschnitt der individuellen Scores für verstopfte Nase, Rhinorrhoe und Niesen) gegenüber dem Ausgangswert.
In der anderen 6-wöchigen Studie wurden SINGULAIR 10 mg (n = 626), Placebo (n = 609) und eine aktive Kontrolle (Cetirizin 10 mg; n = 120) untersucht. Die primäre Analyse verglich die mittlere Veränderung gegenüber dem Ausgangswert des Tages-Scores für nasale Symptome für SINGULAIR vs. Placebo in den ersten 4 Behandlungswochen; Die Studie war nicht auf einen statistischen Vergleich zwischen SINGULAIR und der aktiven Kontrolle ausgelegt. Die primäre Ergebnisvariable umfasste Juckreiz in der Nase zusätzlich zu verstopfter Nase, Rhinorrhoe und Niesen. Der geschätzte Unterschied zwischen SINGULAIR und Placebo betrug -0,04 mit einem 95 %-KI von (-0,09; 0,01). Der geschätzte Unterschied zwischen der aktiven Kontrolle und dem Placebo betrug -0,10 mit einem 95 %-KI von (-0,19, -0,01).
INFORMATIONEN ZUM PATIENTEN
SINGULAIR® (SING-u-lair) (Montelukast-Natrium) Kautabletten
Was sind die wichtigsten Informationen, die ich über SINGULAIR 4 mg wissen sollte?Bei Personen, die SINGULAIR einnahmen, sind schwerwiegende psychische Gesundheitsprobleme aufgetreten oder sogar nach Beendigung der Behandlung.
Dies kann bei Menschen mit oder ohne Vorgeschichte von psychischen Gesundheitsproblemen passieren. Brechen Sie die Einnahme von SINGULAIR ab und informieren Sie unverzüglich Ihren Arzt, wenn Sie oder Ihr Kind ungewöhnliche Verhaltens- oder Denkänderungen feststellen, einschließlich eines der folgenden Symptome:
Was ist SINGULAIR 10mg?
SINGULAIR 4 mg ist ein verschreibungspflichtiges Arzneimittel, das Substanzen im Körper blockiert, die als Leukotriene bezeichnet werden. Dies kann helfen, die Symptome von Asthma und Entzündungen der Nasenschleimhaut (allergische Rhinitis) zu verbessern. SINGULAIR enthält kein Steroid. SINGULAIR wird verwendet für:
Unterlassen Sie Nehmen Sie SINGULAIR ein, wenn Sie gegen einen der Inhaltsstoffe allergisch sind. Eine vollständige Liste der Inhaltsstoffe von SINGULAIR finden Sie am Ende dieses Arzneimittelleitfadens.
Informieren Sie vor der Einnahme von SINGULAIR 4 mg Ihren Arzt über alle Ihre Erkrankungen, auch wenn Sie:
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und pflanzlicher Nahrungsergänzungsmittel. Einige Arzneimittel können die Wirkung von SINGULAIR beeinflussen, oder SINGULAIR 4 mg kann die Wirkung Ihrer anderen Arzneimittel beeinflussen.
Wie sollte ich SINGULAIR 10 mg einnehmen?
Zum jeder Wer nimmt SINGULAIR:
Für Erwachsene und Kinder ab 12 Monaten mit Asthma:
Für Personen ab 6 Jahren zur Vorbeugung von Belastungsasthma:
Für Personen ab 2 Jahren mit saisonaler allergischer Rhinitis oder für Personen ab 6 Monaten mit ganzjähriger allergischer Rhinitis:
Was sollte ich während der Einnahme von SINGULAIR 10 mg vermeiden?
Wenn Sie Asthma haben und Aspirin Ihre Asthmasymptome verschlimmert, vermeiden Sie weiterhin die Einnahme von Aspirin oder anderen Arzneimitteln, die als nichtsteroidale Antirheumatika (NSAIDs) bezeichnet werden, während Sie SINGULAIR einnehmen.
Welche Nebenwirkungen kann SINGULAIR haben?
SINGULAIR kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Informieren Sie sofort Ihren Arzt, wenn Sie eines oder mehrere dieser Symptome bemerken:
Zu den häufigsten Nebenwirkungen von SINGULAIR gehören:
Dies sind nicht alle möglichen Nebenwirkungen von SINGULAIR. Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist SINGULAIR aufzubewahren?
Allgemeine Informationen zur sicheren und wirksamen Anwendung von SINGULAIR.
Medikamente werden manchmal für andere als die in einem Medikationsleitfaden aufgeführten Zwecke verschrieben. Verwenden Sie SINGULAIR nicht für einen Zustand, für den es nicht verschrieben wurde. Geben Sie SINGULAIR nicht an andere Personen weiter, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Sie können Ihren Apotheker oder Gesundheitsdienstleister nach Informationen zu SINGULAIR 10 mg fragen, die für Angehörige der Gesundheitsberufe bestimmt sind.
Was sind die Inhaltsstoffe von SINGULAIR?
Wirkstoff: Montelukast-Natrium
Inaktive Zutaten:
Gebrauchsanweisung
SINGULAIR® (SING-u-lair)(Montelukast-Natrium) Granulat zum Einnehmen
Diese Gebrauchsanweisung enthält Informationen zur Anwendung von SINGULAIR 10 mg Granulat zum Einnehmen.
Wichtige Informationen:
Wie kann ich meinem Kind SINGULAIR 4 mg Granulat zum Einnehmen geben?
Wie ist SINGULAIR 5 mg aufzubewahren?
Diese Gebrauchsanweisung wurde von der US Food and Drug Administration genehmigt
Was ist Skelaxin und wie wird es angewendet?
Skelaxin 400 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Schmerzen des Bewegungsapparates. Skelaxin 400 mg kann allein oder zusammen mit anderen Medikamenten verwendet werden.
Welche Nebenwirkungen kann Skelaxin 400 mg haben?
Skelaxin 400 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Skelaxin gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Skelaxin. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
SKELAXIN® (Metaxalon) ist als ovale 800-mg-Tablette mit rosafarbener Bruchkerbe erhältlich.
Metaxalon ist chemisch gesehen 5-[(3,5-Dimethylphenoxy)methyl]-2-oxazolidinon. Die Summenformel lautet C12H15NO3, was einem Molekulargewicht von 221,25 entspricht. Die Strukturformel lautet:
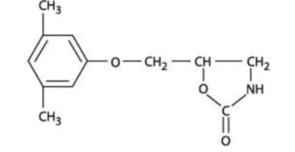
Metaxalon ist ein weißes bis fast weißes, geruchloses kristallines Pulver, das in Chloroform frei löslich ist, in Methanol und in 96 % Ethanol löslich ist, aber in Ether oder Wasser praktisch unlöslich ist.
Jede Tablette enthält 800 mg Metaxalon und die folgenden Hilfsstoffe: Alginsäure, Ammonium-Calcium-Alginat, B-Rose-Flüssigkeit, Maisstärke und Magnesiumstearat.
INDIKATIONEN
SKELAXIN (Metaxalon) ist als Ergänzung zu Ruhe, Physiotherapie und anderen Maßnahmen zur Linderung von Beschwerden in Verbindung mit akuten, schmerzhaften Erkrankungen des Bewegungsapparates indiziert. Die Wirkungsweise dieses Medikaments wurde nicht eindeutig identifiziert, kann aber mit seinen beruhigenden Eigenschaften zusammenhängen. Metaxalone entspannt verspannte Skelettmuskeln beim Menschen nicht direkt.
DOSIERUNG UND ANWENDUNG
Die empfohlene Dosis für Erwachsene und Kinder über 12 Jahren beträgt drei- bis viermal täglich eine Tablette zu 800 mg.
WIE GELIEFERT
SKELAXIN (Metaxalon) ist als ovale 800-mg-Tablette mit rosafarbener Bruchkerbe mit der Prägung „8667“ auf der Bruchkerbe und „S“ auf der anderen Seite erhältlich. Erhältlich in Flaschen zu 100 ( NDC 60793-136-01) und in Flaschen zu 500 ( NDC 60793-136-05).
Bei kontrollierter Raumtemperatur zwischen 15 °C und 30 °C (59 °F und 86 °F) lagern.
SKELAXIN 400 mg ist eine eingetragene Marke von King Pharmaceuticals Research and Development, Inc.
Vertrieb durch: Pfizer Inc., New York, NY 10017. Überarbeitet: November 2015
NEBENWIRKUNGEN
Die häufigsten Reaktionen auf Metaxalon sind:
ZNS: Schläfrigkeit, Schwindel, Kopfschmerzen und Nervosität oder „Reizbarkeit“;
Verdauungs: Übelkeit, Erbrechen, Magen-Darm-Störungen.
Andere Nebenwirkungen sind:
Immunsystem: Überempfindlichkeitsreaktion, Hautausschlag mit oder ohne Juckreiz;
Hämatologisch: Leukopenie; hämolytische Anämie;
Hepatobiliär: Gelbsucht.
Obwohl selten, wurden anaphylaktoide Reaktionen mit Metaxalon berichtet.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Keine Informationen bereitgestellt.
WARNUNGEN
Bei der Anwendung von Metaxalon wurde über ein potenziell lebensbedrohliches Serotonin-Syndrom (SS) berichtet. Diese Berichte traten im Allgemeinen auf, wenn Metaxalon gleichzeitig mit serotonergen Arzneimitteln (wie Tramadol oder selektiven Serotonin-Wiederaufnahmehemmern (SSRIs)) angewendet wurde oder wenn Metaxalon in Dosen angewendet wurde, die höher als die empfohlene Dosis waren (siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und ÜBERDOSIERUNG ). Anzeichen von SS können Klonus, Agitiertheit, Diaphorese, Tremor, Hyperreflexie, Hypertonie und Temperaturerhöhung sein.
SKELAXIN 400 mg kann die Wirkung von Alkohol und anderen zentral dämpfenden Mitteln verstärken.
VORSICHTSMASSNAHMEN
Metaxalon sollte Patienten mit vorbestehender Leberschädigung mit großer Vorsicht verabreicht werden. Bei diesen Patienten sollten serielle Leberfunktionsstudien durchgeführt werden.
Es wurden falsch positive Benedict-Tests aufgrund einer unbekannten reduzierenden Substanz festgestellt. Ein glukosespezifischer Test differenziert die Befunde.
Die Einnahme von SKELAXIN 400 mg zusammen mit Nahrung kann die allgemeine ZNS-Depression verstärken; ältere Patienten können besonders anfällig für diese ZNS-Wirkung sein. (Sehen KLINISCHE PHARMAKOLOGIE : Pharmakokinetik und INFORMATIONEN ZUM PATIENTEN ).
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Das kanzerogene Potenzial von Metaxalon wurde nicht bestimmt.
Schwangerschaft
Reproduktionsstudien an Ratten haben keine Hinweise auf eine Beeinträchtigung der Fertilität oder eine Schädigung des Fötus durch Metaxalon ergeben. Erfahrungen nach der Markteinführung haben keine Hinweise auf eine fötale Verletzung ergeben, aber solche Erfahrungen können die Möglichkeit einer seltenen oder geringfügigen Schädigung des menschlichen Fötus nicht ausschließen. Die sichere Anwendung von Metaxalon wurde im Hinblick auf mögliche Nebenwirkungen auf die fötale Entwicklung nicht nachgewiesen. Daher sollten Metaxalon-Tabletten bei Frauen, die schwanger sind oder werden könnten, und insbesondere während der Frühschwangerschaft, nicht angewendet werden, es sei denn, nach Einschätzung des Arztes überwiegt der potenzielle Nutzen die möglichen Gefahren.
Stillende Mutter
Es ist nicht bekannt, ob dieses Arzneimittel in die Muttermilch übergeht. In der Regel sollte nicht gestillt werden, während ein Patient ein Medikament einnimmt, da viele Medikamente in die Muttermilch ausgeschieden werden.
Pädiatrische Verwendung
Sicherheit und Wirksamkeit bei Kindern im Alter von 12 Jahren und darunter wurden nicht nachgewiesen.
ÜBERDOSIS
Todesfälle durch absichtliche oder versehentliche Überdosierung sind mit Metaxalon aufgetreten, insbesondere in Kombination mit Antidepressiva, und es wurde mit dieser Arzneimittelklasse in Kombination mit Alkohol berichtet.
Ein Serotoninsyndrom wurde berichtet, wenn Metaxalon in Dosen angewendet wurde, die höher als die empfohlene Dosis waren (siehe WARNUNGEN ).
Bei der Bestimmung der LD50 bei Ratten und Mäusen wurden mit steigender Dosierung fortschreitende Sedierung, Hypnose und schließlich Atemversagen festgestellt. Bei Hunden konnte keine LD50 bestimmt werden, da die höheren Dosen innerhalb von 15 bis 30 Minuten eine emetische Wirkung hervorriefen.
Behandlung
Magenspülung und unterstützende Therapie. Es wird empfohlen, sich an ein regionales Giftinformationszentrum zu wenden.
KONTRAINDIKATIONEN
Bekannte Überempfindlichkeit gegen Bestandteile dieses Produkts.
Bekannte Neigung zu arzneimittelinduzierter, hämolytischer oder anderer Anämie.
Signifikant eingeschränkte Nieren- oder Leberfunktion.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Der Wirkungsmechanismus von Metaxalon beim Menschen ist nicht bekannt, kann aber auf eine allgemeine Depression des Zentralnervensystems zurückzuführen sein.
Metaxalone hat keine direkte Wirkung auf den kontraktilen Mechanismus der quergestreiften Muskulatur, der motorischen Endplatte oder der Nervenfaser.
Pharmakokinetik
Die Pharmakokinetik von Metaxalon wurde an gesunden erwachsenen Freiwilligen nach Gabe einer Einzeldosis von SKELAXIN 400 mg unter nüchternen und ernährten Bedingungen in Dosen von 400 mg bis 800 mg untersucht.
Absorption
Maximale Plasmakonzentrationen von Metaxalon treten etwa 3 Stunden nach einer oralen Dosis von 400 mg unter nüchternen Bedingungen auf. Danach nehmen die Metaxalon-Konzentrationen log-linear mit einer terminalen Halbwertszeit von 9,0 ± 4,8 Stunden ab. Die Verdopplung der Dosis von SKELAXIN 400 mg von 400 mg auf 800 mg führt zu einem ungefähr proportionalen Anstieg der Metaxalon-Exposition, wie durch die maximalen Plasmakonzentrationen (Cmax) und die Fläche unter der Kurve (AUC) angezeigt. Die Dosisproportionalität bei Dosen über 800 mg wurde nicht untersucht. Die absolute Bioverfügbarkeit von Metaxalon ist nicht bekannt.
Die pharmakokinetischen Parameter einer Einzeldosis von Metaxalon in zwei Gruppen gesunder Probanden sind in Tabelle 1 dargestellt.
Auswirkungen auf Lebensmittel
Eine randomisierte Zweiweg-Crossover-Studie wurde mit 42 gesunden Freiwilligen (31 Männer, 11 Frauen) durchgeführt, denen eine 400-mg-Tablette SKELAXIN 400 mg im nüchternen Zustand und nach einem fettreichen Standardfrühstück verabreicht wurde. Die Probanden waren zwischen 18 und 48 Jahre alt (Durchschnittsalter = 23,5 ± 5,7 Jahre). Im Vergleich zu nüchternen Bedingungen erhöhte das Vorhandensein einer fettreichen Mahlzeit zum Zeitpunkt der Arzneimittelverabreichung die Cmax um 177,5 % und die AUC (AUC0-t, AUC∞) um 123,5 % bzw. 115,4 %. Auch die Zeit bis zum Erreichen der maximalen Konzentration (Tmax) war nach Nahrungsaufnahme im Vergleich zum nüchternen Zustand verzögert (4,3 h gegenüber 3,3 h) und die terminale Halbwertszeit verkürzt (2,4 h gegenüber 9,0 h).
In einer zweiten Lebensmittelwirkungsstudie mit ähnlichem Design wurden zwei 400-mg-SKELAXIN-400-mg-Tabletten (800 mg) gesunden Freiwilligen (N = 59, 37 Männer, 22 Frauen) im Alter von 18 bis 50 Jahren (Durchschnittsalter = 25,6 ± 8,7 Jahre). Im Vergleich zu nüchternen Bedingungen erhöhte das Vorhandensein einer fettreichen Mahlzeit zum Zeitpunkt der Arzneimittelverabreichung die Cmax um 193,6 % und die AUC (AUC0-t, AUC∞) um 146,4 % bzw. 142,2 %. Die Zeit bis zum Erreichen der maximalen Konzentration (Tmax) war unter Nahrungsaufnahme im Vergleich zu nüchternen Bedingungen ebenfalls verzögert (4,9 h gegenüber 3,0 h) und die terminale Halbwertszeit verkürzt (4,2 h gegenüber 8,0 h). Ähnliche Ergebnisse zu Auswirkungen von Nahrungsmitteln wurden in der oben genannten Studie beobachtet, als eine SKELAXIN 800 mg-Tablette anstelle von zwei SKELAXIN 400 mg-Tabletten verabreicht wurde. Die Zunahme der Metaxalon-Exposition, die mit einer Verkürzung der Halbwertszeit zusammenfällt, kann einer vollständigeren Resorption von Metaxalon in Gegenwart einer fettreichen Mahlzeit zugeschrieben werden (Abbildung 1).
Abbildung 1: Mittlere (SD) Konzentrationen von Metaxalone nach einer 800-mg-Dosis unter nüchternen und ernährten Bedingungen 
Verteilung, Stoffwechsel und Ausscheidung
Obwohl die Plasmaproteinbindung und die absolute Bioverfügbarkeit von Metaxalon nicht bekannt sind, deuten das scheinbare Verteilungsvolumen (V/F ~ 800 l) und die Lipophilie (log P = 2,42) von Metaxalon darauf hin, dass das Arzneimittel umfassend in den Geweben verteilt wird. Metaxalon wird von der Leber metabolisiert und als nicht identifizierte Metaboliten mit dem Urin ausgeschieden. Hepatische Cytochrom-P450-Enzyme spielen eine Rolle bei der Metabolisierung von Metaxalon. Insbesondere CYP1A2, CYP2D6, CYP2E1 und CYP3A4 und in geringerem Maße CYP2C8, CYP2C9 und CYP2C19 scheinen Metaxalon zu metabolisieren.
Metaxalone hemmt wichtige CYP-Enzyme wie CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 und CYP3A4 nicht signifikant. Metaxalone induziert in vitro keine wesentlichen CYP-Enzyme wie CYP1A2, CYP2B6 und CYP3A4.
Pharmakokinetik in speziellen Populationen
Das Alter
Die Auswirkungen des Alters auf die Pharmakokinetik von Metaxalon wurden nach einmaliger Einnahme von zwei 400-mg-Tabletten (800 mg) im nüchternen und ernährten Zustand bestimmt. Die Ergebnisse wurden separat sowie in Kombination mit den Ergebnissen aus drei anderen Studien analysiert. Unter Verwendung der kombinierten Daten weisen die Ergebnisse darauf hin, dass die Pharmakokinetik von Metaxalon unter nüchternen Bedingungen signifikant stärker vom Alter beeinflusst wird als unter nüchternen Bedingungen, wobei die Bioverfügbarkeit unter nüchternen Bedingungen mit zunehmendem Alter zunimmt.
Die Bioverfügbarkeit von Metaxalon unter nüchternen und ernährten Bedingungen in drei Gruppen gesunder Freiwilliger unterschiedlichen Alters ist in Tabelle 2 gezeigt.
Geschlecht
Die Auswirkung des Geschlechts auf die Pharmakokinetik von Metaxalon wurde in einer offenen Studie untersucht, in der 48 gesunden erwachsenen Freiwilligen (24 Männer, 24 Frauen) zwei SKELAXIN 400-mg-Tabletten (800 mg) im nüchternen Zustand verabreicht wurden. Die Bioverfügbarkeit von Metaxalon war bei Frauen signifikant höher als bei Männern, wie durch Cmax (2115 ng/ml versus 1335 ng/ml) und AUC∞ (17884 ng·h/ml versus 10328 ng·h/ml) belegt wurde. Die mittlere Halbwertszeit betrug 11,1 Stunden bei Frauen und 7,6 Stunden bei Männern. Das scheinbare Verteilungsvolumen von Metaxalon war bei Männern etwa 22 % höher als bei Frauen, unterschied sich jedoch nicht signifikant, wenn es an das Körpergewicht angepasst wurde. Ähnliche Ergebnisse wurden auch gesehen, als der zuvor beschriebene kombinierte Datensatz in der Analyse verwendet wurde.
Leber-/Niereninsuffizienz
Die Auswirkungen von Leber- und Nierenerkrankungen auf die Pharmakokinetik von Metaxalon wurden nicht bestimmt. In Ermangelung solcher Informationen sollte SKELAXIN 400 mg bei Patienten mit eingeschränkter Leber- und/oder Nierenfunktion mit Vorsicht angewendet werden.
INFORMATIONEN ZUM PATIENTEN
SKELAXIN kann die geistigen und/oder körperlichen Fähigkeiten beeinträchtigen, die für die Ausführung gefährlicher Aufgaben wie das Bedienen von Maschinen oder das Führen eines Kraftfahrzeugs erforderlich sind, insbesondere wenn es mit Alkohol oder anderen ZNS-dämpfenden Mitteln eingenommen wird.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Die sedierenden Wirkungen von SKELAXIN und anderen ZNS-dämpfenden Mitteln (z. B. Alkohol, Benzodiazepine, Opioide, trizyklische Antidepressiva) können sich addieren. Daher ist bei Patienten, die gleichzeitig mehr als einen dieser ZNS-Depressiva einnehmen, Vorsicht geboten.
Aufgrund des Potenzials für SS ist Vorsicht geboten, wenn Metaxalon zusammen mit Arzneimitteln verabreicht wird, die das serotonerge Neurotransmittersystem beeinflussen können, wie Tramadol oder SSRIs (siehe WARNUNGEN ).
Was ist Spiriva 9 mcg HandiHaler und wie wird es angewendet?
Spiriva HandiHaler (Tiotropiumbromid) Inhalationspulver ist ein Anticholinergikum, das zur Vorbeugung von Bronchospasmus (Verengung der Atemwege in der Lunge) bei Menschen mit Bronchitis, Emphysem oder COPD (chronisch obstruktive Lungenerkrankung) angewendet wird.
Welche Nebenwirkungen hat Spiriva 9mcg HandiHaler?
Häufige Nebenwirkungen von Spiriva 9 mcg HandiHaler sind:
Informieren Sie Ihren Arzt, wenn Sie schwerwiegende Nebenwirkungen von Spiriva 9 mcg HandiHaler haben, einschließlich:
BEZEICHNUNG
SPIRIVA 9mcg HANDIHALER besteht aus SPIRIVA-Kapseln und einem HANDIHALER-Gerät. Jede hellgrüne SPIRIVA-Hartgelatinekapsel 9 µg enthält ein Trockenpulver bestehend aus 18 µg Tiotropium (entsprechend 22,5 µg Tiotropiumbromid-Monohydrat), gemischt mit Lactose-Monohydrat (das Milchproteine enthalten kann).
Der Inhalt der SPIRIVA-Kapseln ist nur zur oralen Inhalation und nur zur Verabreichung mit dem HANDIHALER-Gerät bestimmt.
Der Wirkstoff von SPIRIVA HANDIHALER ist Tiotropium. Der Wirkstoff, Tiotropiumbromid-Monohydrat, ist ein Anticholinergikum mit Spezifität für Muskarinrezeptoren. Es wird chemisch beschrieben als (1α, 2β, 4β, 5α, 7β)-7-[(Hydroxydi-2-thienylacetyl)oxy]-9,9-dimethyl-3-oxa-9-azoniatricyclo[3.3.1.02,4] Nonanbromidmonohydrat. Es ist eine synthetische, nicht-chirale, quartäre Ammoniumverbindung. Tiotropiumbromid ist ein weißes oder gelblich weißes Pulver. Es ist schwer löslich in Wasser und löslich in Methanol.
Die Strukturformel lautet:
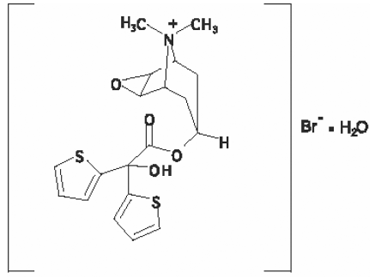
Tiotropiumbromid (Monohydrat) hat eine Molekülmasse von 490,4 und eine Summenformel von C19H22NO4S2Br • H2O.
Das HANDIHALER-Gerät ist ein Inhalationsgerät zum Inhalieren des Trockenpulvers, das in der SPIRIVA 9-mcg-Kapsel enthalten ist. Das Trockenpulver wird vom HANDIHALER-Gerät mit Durchflussraten von nur 20 l/min abgegeben. Bei standardisierten In-vitro-Tests gibt das HANDIHALER-Gerät durchschnittlich 10,4 mcg Tiotropium ab, wenn es bei einer Flussrate von 39 l/min für 3,1 Sekunden (insgesamt 2 l) getestet wird. In einer Studie mit 26 erwachsenen Patienten mit COPD und stark eingeschränkter Lungenfunktion [mittleres FEV1 1,02 l (Bereich 0,45 bis 2,24 l); 37,6 % des Sollwerts (Bereich 16 % bis 65 %)] betrug der mediane Spitzeninspirationsfluss (PIF) durch das HANDIHALER-Gerät 30,0 l/min (Bereich 20,4 bis 45,6 l/min). Die Menge des an die Lunge abgegebenen Arzneimittels hängt von Faktoren des Patienten ab, wie z. B. Inspirationsfluss und maximaler Inspirationsfluss durch das HANDIHALER-Gerät, die von Patient zu Patient und mit der Expositionszeit der SPIRIVA 9-mcg-Kapsel außerhalb der Blisterpackung variieren können Pack.
INDIKATIONEN
SPIRIVA HANDIHALER (Tiotropiumbromid-Pulver zur Inhalation) ist indiziert zur einmal täglichen Langzeitbehandlung von Bronchospasmen im Zusammenhang mit chronisch obstruktiver Lungenerkrankung (COPD), einschließlich chronischer Bronchitis und Emphysem. SPIRIVA 9mcg HANDIHALER ist angezeigt, um Exazerbationen bei COPD-Patienten zu reduzieren.
DOSIERUNG UND ANWENDUNG
Nur zur oralen Inhalation. Schlucken Sie SPIRIVA 9 mcg Kapseln nicht, da die beabsichtigte Wirkung auf die Lunge nicht erreicht wird. Der Inhalt der SPIRIVA-Kapseln sollte nur mit dem HANDIHALER-Gerät verwendet werden [sehen ÜBERDOSIS ].
Die empfohlene Dosis von SPIRIVA HANDIHALER besteht aus zwei Inhalationen des Pulverinhalts einer SPIRIVA-Kapsel einmal täglich mit dem HANDIHALER-Gerät [siehe INFORMATIONEN ZUM PATIENTEN ]. Nehmen Sie nicht mehr als eine Dosis in 24 Stunden ein.
Zur Verabreichung von SPIRIVA HANDIHALER wird eine 9-mcg-SPIRIVA-Kapsel in die mittlere Kammer des HANDIHALER-Geräts gegeben. Die SPIRIVA 9mcg-Kapsel wird durch Drücken und Loslassen der grünen Anstechtaste an der Seite des HANDIHALER-Geräts durchstochen. Die Tiotropium-Formulierung wird im Luftstrom dispergiert, wenn der Patient durch das Mundstück einatmet [siehe INFORMATIONEN ZUM PATIENTEN ].
Bei geriatrischen, hepatisch oder nierenbeeinträchtigten Patienten ist keine Dosisanpassung erforderlich. Patienten mit mäßiger bis schwerer Nierenfunktionsstörung, die SPIRIVA 9 µg HANDIHALER erhalten, sollten jedoch engmaschig auf anticholinerge Wirkungen überwacht werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , Verwendung in bestimmten Bevölkerungsgruppen , und KLINISCHE PHARMAKOLOGIE ].
WIE GELIEFERT
Darreichungsformen und Stärken
Pulver zur Inhalation
SPIRIVA 9 µg HANDIHALER besteht aus SPIRIVA-Kapseln mit Tiotropium-Pulver zur oralen Inhalation und einem HANDIHALER-Gerät. SPIRIVA 9 µg Kapseln enthalten 18 µg Tiotropium in einer hellgrünen Hartgelatinekapsel mit dem Aufdruck TI 01 auf einer Seite und dem Firmenlogo von Boehringer Ingelheim auf der anderen Seite. Das HANDIHALER-Gerät ist nur für die Verwendung mit den SPIRIVA-Kapseln vorgesehen.
Lagerung und Handhabung
SPIRIVA HANDIHALER besteht aus SPIRIVA 9mcg-Kapseln und dem HANDIHALER-Gerät. SPIRIVA 9 µg Kapseln enthalten 18 µg Tiotropium und sind hellgrün, mit dem Firmenlogo von Boehringer Ingelheim auf der SPIRIVA Kapselkappe und TI 01 auf dem SPIRIVA Kapselunterteil oder umgekehrt.
Das HANDIHALER-Gerät ist grau mit einem grünen Durchstechknopf. Es ist mit SPIRIVA HANDIHALER (Tiotropiumbromid-Pulver zur Inhalation), dem Firmenlogo von Boehringer Ingelheim, bedruckt. Es ist auch aufgedruckt, um darauf hinzuweisen, dass SPIRIVA-Kapseln nicht im HANDIHALER-Gerät aufbewahrt werden sollten und dass das HANDIHALER-Gerät nur mit SPIRIVA-9-mcg-Kapseln verwendet werden darf.
SPIRIVA 9 µg Kapseln sind in einer Aluminium/Aluminium-Blisterkarte verpackt und entlang einer perforierten Schnittlinie verbunden. SPIRIVA 9 mcg Kapseln sollten immer in der Blisterpackung aufbewahrt und erst unmittelbar vor der Anwendung entnommen werden. Das Medikament sollte sofort verwendet werden, nachdem die Verpackung über einer einzelnen SPIRIVA-Kapsel geöffnet wurde.
Folgende Pakete sind verfügbar:
Außerhalb der Reichweite von Kindern aufbewahren. Puder nicht in die Augen bringen.
Lagerung
Bei 25 °C (77 °F) lagern; Abweichungen bis 15° bis 30°C (59° bis 86°F) zulässig [siehe USP Controlled Room Temperature].
Die SPIRIVA-Kapseln sollten keinen extremen Temperaturen oder Feuchtigkeit ausgesetzt werden. Bewahren Sie SPIRIVA 9 mcg-Kapseln nicht im HANDIHALER-Gerät auf.
Vertrieb durch: Boehringer Ingelheim Pharmaceuticals, Inc. Ridgefield, CT 06877 USA. Überarbeitet: Februar 2018
NEBENWIRKUNGEN
Die folgenden Nebenwirkungen werden in anderen Abschnitten beschrieben oder ausführlicher beschrieben:
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, kann die Inzidenz von Nebenwirkungen, die in klinischen Studien mit einem Medikament beobachtet wurden, nicht direkt mit der Inzidenz in klinischen Studien mit einem anderen Medikament verglichen werden und spiegelt möglicherweise nicht die in der Praxis beobachteten Inzidenzen wider.
6-monatige bis 1-jährige Testversionen
Die unten beschriebenen Daten spiegeln die Exposition gegenüber SPIRIVA 9 mcg HANDIHALER bei 2663 Patienten wider. SPIRIVA 9mcg HANDIHALER wurde in zwei 1-jährigen placebokontrollierten Studien, zwei 1-jährigen aktiv-kontrollierten Studien und zwei 6-monatigen placebokontrollierten Studien bei Patienten mit COPD untersucht. In diesen Studien wurden 1308 Patienten mit SPIRIVA HANDIHALER in der empfohlenen Dosis von 18 µg einmal täglich behandelt. Die Bevölkerung war zwischen 39 und 87 Jahre alt, wobei 65 % bis 85 % Männer, 95 % Kaukasier waren, und COPD mit einem mittleren prognostizierten FEV1-Prozentsatz von 39 % bis 43 % hatte. Patienten mit Engwinkelglaukom oder symptomatischer Prostatahypertrophie oder Obstruktion des Blasenausgangs wurden von diesen Studien ausgeschlossen. Eine zusätzliche 6-monatige Studie, die in einer Umgebung für Veteranenangelegenheiten durchgeführt wurde, ist in dieser Sicherheitsdatenbank nicht enthalten, da nur schwerwiegende unerwünschte Ereignisse erfasst wurden.
Die am häufigsten berichtete Nebenwirkung war Mundtrockenheit. Mundtrockenheit war in der Regel leicht und verschwand oft während der fortgesetzten Behandlung. Andere bei einzelnen Patienten berichtete Reaktionen, die mit möglichen anticholinergen Wirkungen vereinbar sind, waren Verstopfung, Tachykardie, verschwommenes Sehen, Glaukom (neues Auftreten oder Verschlechterung), Dysurie und Harnverhalt.
Vier multizentrische placebokontrollierte und aktivkontrollierte Einjahresstudien untersuchten SPIRIVA HANDIHALER bei Patienten mit COPD. Tabelle 1 zeigt alle Nebenwirkungen, die mit einer Häufigkeit von ≥ 3 % in der SPIRIVA 9 mcg HANDIHALER-Gruppe in den placebokontrollierten 1-Jahres-Studien auftraten, in denen die Raten in der SPIRIVA HANDIHALER-Gruppe um ≥ 1 % über Placebo lagen. Zum Vergleich ist die Häufigkeit entsprechender Reaktionen in den Ipratropium-kontrollierten Studien enthalten.
Arthritis, Husten und grippeähnliche Symptome traten in der Behandlungsgruppe mit SPIRIVA 9 mcg HANDIHALER mit einer Rate von ≥ 3 % auf, waren jedoch
Andere Reaktionen, die in der SPIRIVA HANDIHALER-Gruppe mit einer Häufigkeit von 1 % bis 3 % in den Placebo-kontrollierten Studien auftraten, in denen die Raten höher waren als in der Placebo-Gruppe, sind:
Körper als Ganzes: allergische Reaktion, Beinschmerzen;
Zentrales und peripheres Nervensystem: Dysphonie, Parästhesien;
Erkrankungen des Magen-Darm-Trakts: gastrointestinale Störung nicht anders angegeben (NOS), gastroösophagealer Reflux, Stomatitis (einschließlich ulzerativer Stomatitis);
Stoffwechsel- und Ernährungsstörungen: Hypercholesterinämie, Hyperglykämie;
Erkrankungen des Bewegungsapparates: Skelettschmerzen;
Herzereignisse: Angina pectoris (einschließlich verschlimmerter Angina pectoris);
Psychische Störung: Depression; Infektionen: Herpes zoster;
Erkrankung des Atmungssystems (oben): Laryngitis;
Sehstörung: Katarakt.
Zu den in den klinischen Studien beobachteten Nebenwirkungen mit einer Inzidenz von
In den 1-Jahres-Studien nahm die Inzidenz von Mundtrockenheit, Obstipation und Harnwegsinfektionen mit dem Alter zu [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Zwei multizentrische, 6-monatige, kontrollierte Studien untersuchten SPIRIVA 9 mcg HANDIHALER bei Patienten mit COPD. Die Nebenwirkungen und Inzidenzraten waren ähnlich denen, die in den kontrollierten 1-Jahres-Studien beobachtet wurden.
4-Jahres-Testversion
Die unten beschriebenen Daten spiegeln die Exposition gegenüber SPIRIVA 9 mcg HANDIHALER bei 5992 COPD-Patienten in einer 4-jährigen placebokontrollierten Studie wider. In dieser Studie wurden 2986 Patienten mit SPIRIVA 9 µg HANDIHALER in der empfohlenen Dosis von 18 µg einmal täglich behandelt. Die Population war zwischen 40 und 88 Jahre alt, war zu 75 % männlich, zu 90 % Kaukasier und hatte COPD mit einem erwarteten mittleren FEV1-Prozentsatz vor Bronchodilatation von 40 %. Patienten mit Engwinkelglaukom oder symptomatischer Prostatahypertrophie oder Obstruktion des Blasenausgangs wurden von diesen Studien ausgeschlossen. Als die Nebenwirkungen mit einer Häufigkeit von ≥ 3 % in der SPIRIVA 9 µg HANDIHALER-Gruppe analysiert wurden, wobei die Raten in der SPIRIVA 9 µg HANDIHALER-Gruppe die Placebo-Rate um ≥ 1 % überstiegen, schlossen die Nebenwirkungen ein (SPIRIVA 9 µg HANDIHALER, Placebo): Pharyngitis (12,5 % , 10,8 %), Sinusitis (6,5 %, 5,3 %), Kopfschmerzen (5,7 %, 4,5 %), Verstopfung (5,1 %, 3,7 %), Mundtrockenheit (5,1 %, 2,7 %), Depression (4,4 %, 3,3 %) , Schlaflosigkeit (4,4 %, 3,0 %) und Arthralgie (4,2 %, 3,1 %).
Zusätzliche Nebenwirkungen
Andere bisher nicht aufgeführte Nebenwirkungen, die bei COPD-Patienten, die mit SPIRIVA 9 mcg HANDIHALER behandelt wurden, häufiger berichtet wurden als Placebo, sind: Dehydratation, Hautgeschwür, Stomatitis, Gingivitis, oropharyngeale Candidiasis, trockene Haut, Hautinfektion und Gelenkschwellung.
Postmarketing-Erfahrung
Bei der weltweiten Verwendung von SPIRIVA HANDIHALER nach der Zulassung wurden Nebenwirkungen festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen. Diese Nebenwirkungen sind: Reizungen an der Applikationsstelle (Glossitis, Geschwürbildung im Mund und pharyngolaryngeale Schmerzen), Schwindel, Dysphagie, Heiserkeit, Darmverschluss einschließlich Lähmung des Ileus, erhöhter Augeninnendruck, orale Candidiasis, Herzklopfen, Pruritus, Tachykardie, Rachenreizung und Urtikaria.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Sympathomimetika, Methylxanthine, Steroide
SPIRIVA 9 µg HANDIHALER wurde gleichzeitig mit kurz und lang wirksamen sympathomimetischen (Beta-Agonisten), Bronchodilatatoren, Methylxanthinen und oralen und inhalativen Steroiden ohne Zunahme von Nebenwirkungen angewendet.
Anticholinergika
Es besteht die Möglichkeit einer additiven Wechselwirkung mit gleichzeitig angewendeten Anticholinergika. Vermeiden Sie daher die gleichzeitige Anwendung von SPIRIVA 9 µg HANDIHALER mit anderen Anticholinergika-haltigen Arzneimitteln, da dies zu einer Zunahme von anticholinergen Nebenwirkungen führen kann [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Nicht für den akuten Gebrauch
SPIRIVA 9mcg HANDIHALER ist als einmal tägliche Erhaltungstherapie bei COPD vorgesehen und sollte nicht zur Linderung akuter Symptome, dh als Notfalltherapie zur Behandlung akuter Episoden von Bronchospasmen, angewendet werden.
Sofortige Überempfindlichkeitsreaktionen
Unmittelbare Überempfindlichkeitsreaktionen, einschließlich Urtikaria, Angioödem (einschließlich Schwellung der Lippen, der Zunge oder des Rachens), Hautausschlag, Bronchospasmus, Anaphylaxie oder Juckreiz, können nach Verabreichung von SPIRIVA 9 mcg HANDIHALER auftreten. Wenn eine solche Reaktion auftritt, sollte die Behandlung mit SPIRIVA 9 mcg HANDIHALER sofort abgebrochen und alternative Behandlungen in Betracht gezogen werden. Angesichts der ähnlichen Strukturformel von Atropin und Tiotropium sollten Patienten mit einer Vorgeschichte von Überempfindlichkeitsreaktionen auf Atropin oder seine Derivate engmaschig auf ähnliche Überempfindlichkeitsreaktionen auf SPIRIVA HANDIHALER überwacht werden. Darüber hinaus sollte SPIRIVA HANDIHALER bei Patienten mit schwerer Überempfindlichkeit gegen Milchproteine mit Vorsicht angewendet werden.
Paradoxer Bronchospasmus
Inhalative Arzneimittel, einschließlich SPIRIVA HANDIHALER, können paradoxe Bronchospasmen verursachen. Wenn dies auftritt, sollte es sofort mit einem inhalativen kurz wirksamen Beta2-Agonisten wie Albuterol behandelt werden. Die Behandlung mit SPIRIVA HANDIHALER sollte abgebrochen und andere Behandlungen in Betracht gezogen werden.
Verschlechterung des Engwinkelglaukoms
SPIRIVA 9mcg HANDIHALER sollte bei Patienten mit Engwinkelglaukom mit Vorsicht angewendet werden. Verschreibende Ärzte und Patienten sollten auf Anzeichen und Symptome eines akuten Engwinkelglaukoms achten (z. B. Augenschmerzen oder -beschwerden, verschwommenes Sehen, Lichthöfe oder Farbbilder in Verbindung mit roten Augen aufgrund einer konjunktivalen Kongestion und Hornhautödem). Weisen Sie die Patienten an, sofort einen Arzt aufzusuchen, wenn eines dieser Anzeichen oder Symptome auftritt.
Verschlechterung der Harnverhaltung
SPIRIVA HANDIHALER sollte bei Patienten mit Harnverhalt mit Vorsicht angewendet werden. Verschreibende Ärzte und Patienten sollten auf Anzeichen und Symptome einer Harnverhaltung achten (z. B. Schwierigkeiten beim Wasserlassen, schmerzhaftes Wasserlassen), insbesondere bei Patienten mit Prostatahyperplasie oder Blasenhalsobstruktion. Weisen Sie die Patienten an, sofort einen Arzt aufzusuchen, wenn eines dieser Anzeichen oder Symptome auftritt.
Nierenfunktionsstörung
Da es sich um ein überwiegend renal ausgeschiedenes Arzneimittel handelt, sollten Patienten mit mäßiger bis schwerer Nierenfunktionsstörung (Kreatinin-Clearance KLINISCHE PHARMAKOLOGIE ].
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( PATIENTENINFORMATION und Gebrauchsanweisung ).
Nicht für den akuten Gebrauch
Weisen Sie die Patienten darauf hin, dass SPIRIVA HANDIHALER ein einmal täglich zu verabreichender Erhaltungs-Bronchodilatator ist und nicht zur sofortigen Linderung von Atemproblemen (dh als Notfallmedikation) verwendet werden sollte.
Sofortige Überempfindlichkeitsreaktionen
Informieren Sie die Patienten darüber, dass nach der Verabreichung von SPIRIVA 9 mcg HANDIHALER Anaphylaxie, Angioödem (einschließlich Schwellung der Lippen, der Zunge oder des Rachens), Urtikaria, Hautausschlag, Bronchospasmus oder Juckreiz auftreten können. Weisen Sie den Patienten an, die Behandlung sofort abzubrechen und einen Arzt aufzusuchen, wenn eines dieser Anzeichen oder Symptome auftritt.
Paradoxer Bronchospasmus
Informieren Sie die Patienten darüber, dass SPIRIVA HANDIHALER paradoxe Bronchospasmen hervorrufen kann. Weisen Sie die Patienten darauf hin, dass sie SPIRIVA HANDIHALER absetzen sollten, wenn ein paradoxer Bronchospasmus auftritt.
Verschlechterung des Engwinkelglaukoms
Weisen Sie die Patienten an, auf Anzeichen und Symptome eines Engwinkelglaukoms zu achten (z. B. Augenschmerzen oder -beschwerden, verschwommenes Sehen, Lichthöfe oder Farbbilder in Verbindung mit roten Augen aufgrund einer konjunktivalen Verstopfung und eines Hornhautödems). Weisen Sie die Patienten an, sofort einen Arzt aufzusuchen, wenn eines dieser Anzeichen und Symptome auftritt.
Informieren Sie die Patienten darüber, dass darauf geachtet werden muss, dass das Pulver nicht in die Augen gelangt, da dies zu verschwommenem Sehen und erweiterten Pupillen führen kann.
Da bei der Anwendung von SPIRIVA HANDIHALER Schwindel und verschwommenes Sehen auftreten können, sollten Patienten davor gewarnt werden, sich an Aktivitäten wie dem Führen eines Fahrzeugs oder dem Bedienen von Geräten oder Maschinen zu beteiligen.
Verschlechterung der Harnverhaltung
Weisen Sie die Patienten an, auf Anzeichen und Symptome einer Harnverhaltung (z. B. Schwierigkeiten beim Wasserlassen, Schmerzen beim Wasserlassen) zu achten. Weisen Sie die Patienten an, sofort einen Arzt aufzusuchen, wenn eines dieser Anzeichen oder Symptome auftritt.
Anweisungen zur Verabreichung von SPIRIVA HANDIHALER
Unterweisen Sie die Patienten in der korrekten Verabreichung von SPIRIVA 9 mcg-Kapseln mit dem HANDIHALER-Gerät [siehe INFORMATIONEN ZUM PATIENTEN ]. Weisen Sie die Patienten darauf hin, dass SPIRIVA 9 mcg-Kapseln nur über das HANDIHALER-Gerät verabreicht werden sollten und das HANDIHALER-Gerät nicht zur Verabreichung anderer Medikamente verwendet werden sollte. Erinnern Sie die Patienten daran, dass der Inhalt von SPIRIVA 9 mcg-Kapseln nur zur oralen Inhalation bestimmt ist und nicht geschluckt werden darf.
Weisen Sie die Patienten an, SPIRIVA 9 mcg-Kapseln immer in versiegelten Blisterpackungen aufzubewahren und unmittelbar vor der Anwendung nur eine SPIRIVA-Kapsel zu entnehmen, da sonst die Wirksamkeit verringert werden kann. Weisen Sie die Patienten an, unbenutzte zusätzliche SPIRIVA-Kapseln, die der Luft ausgesetzt waren (dh nicht für den sofortigen Gebrauch bestimmt sind), zu entsorgen.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
In einer 104-wöchigen Inhalationsstudie an Ratten mit Tiotropium-Dosen von bis zu 59 µg/kg/Tag, in einer 83-wöchigen Inhalationsstudie an weiblichen Mäusen mit Dosen von bis zu 145 µg/kg/Tag und in eine 101-wöchige Inhalationsstudie an männlichen Mäusen mit Dosen von bis zu 2 mcg/kg/Tag. Diese Dosen entsprechen etwa dem 30-, 40- bzw. 0,5-fachen der empfohlenen täglichen Inhalationsdosis beim Menschen (MRHDID) auf µg/m2-Basis.
Tiotropiumbromid zeigte in den folgenden Assays keine Hinweise auf Mutagenität oder Klastogenität: dem bakteriellen Genmutationsassay, dem V79-Zellmutageneseassay des chinesischen Hamsters, den Chromosomenaberrationsassays in humanen Lymphozyten in vitro und der Bildung von Mikrokernen in der Maus in vivo sowie der außerplanmäßigen DNA-Synthese in primärer Rattenhepatozyten-in-vitro-Assay.
Bei Ratten wurde bei inhalativen Tiotropium-Dosen von 78 µg/kg/Tag oder mehr (etwa das 40-fache der MRHDID auf µg/m2-Basis) eine Abnahme der Anzahl der Corpora lutea und des Prozentsatzes der Implantate festgestellt. Bei 9 µg/kg/Tag (ungefähr das 5-fache der MRHDID auf µg/m2-Basis) wurden keine derartigen Wirkungen beobachtet. Der Fertilitätsindex wurde jedoch bei Inhalationsdosen von bis zu 1689 µg/kg/Tag (ungefähr das 910-fache der MRHDID auf µg/m2-Basis) nicht beeinflusst.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Zusammenfassung der Risiken
Die begrenzten Humandaten zur Anwendung von SPIRIVA 9mcg HANDIHALER während der Schwangerschaft reichen nicht aus, um über ein arzneimittelbedingtes Risiko unerwünschter schwangerschaftsbedingter Folgen zu informieren. Basierend auf Reproduktionsstudien an Tieren wurden keine strukturellen Anomalien beobachtet, wenn Tiotropium trächtigen Ratten und Kaninchen während der Organogenese in Dosen verabreicht wurde, die das 790- bzw. 8-fache der maximal empfohlenen täglichen Inhalationsdosis beim Menschen (MRHDID) betrugen. Bei Ratten und Kaninchen, denen Tiotropium in für das Muttertier toxischen Dosen verabreicht wurde, die das 430-Fache bzw. das 40-Fache der MRHDID betrugen, wurde ein erhöhter Verlust nach der Implantation beobachtet [vgl Daten ].
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten für die angegebene Population ist unbekannt. Alle Schwangerschaften haben ein Hintergrundrisiko für Geburtsfehler, Verlust oder andere unerwünschte Folgen. In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwere Geburtsfehler und Fehlgeburten bei klinisch erkannten Schwangerschaften bei 2 % bis 4 % bzw. 15 % bis 20 %.
Daten
Tierdaten
In 2 getrennten Studien zur embryofetalen Entwicklung erhielten trächtige Ratten und Kaninchen Tiotropium während der Organogenese in Dosen bis zu etwa dem 790- bzw. 8-fachen der MRHDID (auf Mikrogramm/m2-Basis bei Inhalationsdosen von 1471 und 7 Mikrogramm/kg). /Tag bei Ratten bzw. Kaninchen). Bei Ratten oder Kaninchen wurden keine Hinweise auf strukturelle Anomalien beobachtet. Bei Ratten verursachte Tiotropium jedoch fötale Resorption, Wurfverlust, eine Abnahme der Anzahl lebender Jungtiere bei der Geburt und des mittleren Gewichts der Jungtiere sowie eine Verzögerung der sexuellen Reifung der Jungtiere bei Tiotropium-Dosen von etwa dem 40-fachen der MRHDID (bei einem mcg/m2 basierend auf einer mütterlichen Inhalationsdosis von 78 mcg/kg/Tag). Bei Kaninchen verursachte Tiotropium bei einer Tiotropium-Dosis von etwa dem 430-Fachen der MRHDID (auf einer mcg/m2-Basis bei einer maternalen Inhalationsdosis von 400 mcg/kg/Tag) einen Anstieg des Postimplantationsverlusts. Solche Wirkungen wurden bei etwa dem 5- bzw. 95-Fachen der MRHDID nicht beobachtet (auf Mikrogramm/m2-Basis bei Inhalationsdosen von 9 bzw. 88 Mikrogramm/kg/Tag bei Ratten bzw. Kaninchen).
Stillzeit
Zusammenfassung der Risiken
Es liegen keine Daten über das Vorhandensein von Tiotropium in der Muttermilch, die Auswirkungen auf den gestillten Säugling oder die Auswirkungen auf die Milchproduktion vor. Tiotropium ist in der Milch säugender Ratten vorhanden; aufgrund artspezifischer Unterschiede in der Laktationsphysiologie ist die klinische Relevanz dieser Daten jedoch nicht klar [vgl Daten ]. Die Entwicklungs- und Gesundheitsvorteile des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an SPIRIVA 9 mcg HANDIHALER und möglichen Nebenwirkungen von SPIRIVA HANDIHALER auf das gestillte Kind oder von der zugrunde liegenden Erkrankung der Mutter berücksichtigt werden.
Daten
Die Verteilung von Tiotropiumbromid in die Milch wurde nach einmaliger intravenöser Verabreichung von 10 mg/kg an laktierende Ratten untersucht. Tiotropium und/oder seine Metaboliten sind in der Milch säugender Ratten in höheren Konzentrationen als im Plasma vorhanden.
Pädiatrische Verwendung
SPIRIVA HANDIHALER ist nicht für die Anwendung bei Kindern angezeigt. Die Sicherheit und Wirksamkeit von SPIRIVA 9mcg HANDIHALER bei pädiatrischen Patienten wurde nicht nachgewiesen.
Geriatrische Verwendung
Basierend auf den verfügbaren Daten ist keine Anpassung der SPIRIVA HANDIHALER-Dosierung bei geriatrischen Patienten gerechtfertigt [siehe KLINISCHE PHARMAKOLOGIE ].
Von der Gesamtzahl der Patienten, die SPIRIVA 9 mcg HANDIHALER in den 1-jährigen klinischen Studien erhielten, waren 426
Nierenfunktionsstörung
Patienten mit mittelschwerer bis schwerer Nierenfunktionsstörung (Kreatinin-Clearance DOSIERUNG UND ANWENDUNG , WARNUNGEN UND VORSICHTSMASSNAHMEN , und KLINISCHE PHARMAKOLOGIE ].
Leberfunktionsstörung
Die Auswirkungen einer Leberfunktionsstörung auf die Pharmakokinetik von Tiotropium wurden nicht untersucht.
ÜBERDOSIS
Hohe Dosen von Tiotropium können zu anticholinergen Anzeichen und Symptomen führen. Allerdings traten bei 6 gesunden Probanden nach einer inhalierten Einzeldosis von bis zu 282 µg Tiotropium keine systemischen anticholinergen Nebenwirkungen auf. In einer Studie mit 12 gesunden Probanden wurden nach wiederholter einmal täglicher Inhalation von 141 µg Tiotropium bilaterale Konjunktivitis und Mundtrockenheit beobachtet.
Die Behandlung einer Überdosierung besteht aus dem Absetzen von SPIRIVA HANDIHALER zusammen mit der Einleitung einer geeigneten symptomatischen und/oder unterstützenden Therapie.
Versehentliche Einnahme
Eine akute Vergiftung durch versehentliche orale Einnahme von SPIRIVA-Kapseln ist unwahrscheinlich, da es systemisch nicht gut resorbiert wird.
Aus Erfahrungen nach Markteinführung wurde ein Fall von Überdosierung berichtet. Es wurde berichtet, dass eine Patientin 30 Kapseln über einen Zeitraum von 2,5 Tagen inhaliert hatte und einen veränderten Geisteszustand, Zittern, Bauchschmerzen und schwere Verstopfung entwickelte. Der Patient wurde ins Krankenhaus eingeliefert, SPIRIVA HANDIHALER wurde abgesetzt und die Verstopfung mit einem Einlauf behandelt. Der Patient erholte sich und wurde noch am selben Tag entlassen.
KONTRAINDIKATIONEN
SPIRIVA 9mcg HANDIHALER ist kontraindiziert bei Patienten mit einer Überempfindlichkeit gegen Tiotropium, Ipratropium oder einen der Bestandteile dieses Produkts [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. In klinischen Studien und Erfahrungen nach der Markteinführung mit SPIRIVA 9 mcg HANDIHALER wurden unmittelbare Überempfindlichkeitsreaktionen, einschließlich Angioödem (einschließlich Schwellung der Lippen, Zunge oder Rachen), Juckreiz oder Hautausschlag berichtet [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Tiotropium ist ein lang wirkendes Antimuskarinikum, das oft als Anticholinergikum bezeichnet wird. Es hat eine ähnliche Affinität zu den Subtypen der Muskarinrezeptoren M1 bis M5. In den Atemwegen zeigt es pharmakologische Wirkungen durch Hemmung von M3-Rezeptoren an der glatten Muskulatur, was zu einer Bronchodilatation führt. Die kompetitive und reversible Natur des Antagonismus wurde mit Rezeptoren menschlichen und tierischen Ursprungs und isolierten Organpräparaten gezeigt. In präklinischen In-vitro- und In-vivo-Studien war die Verhinderung von Methacholin-induzierten Bronchokonstriktionseffekten dosisabhängig und hielt länger als 24 Stunden an. Die Bronchodilatation nach Inhalation von Tiotropium ist überwiegend ein ortsspezifischer Effekt.
Pharmakodynamik
Herzelektrophysiologie
In einer multizentrischen, randomisierten, doppelblinden Studie mit Tiotropium-Trockenpulver zur Inhalation, an der 198 Patienten mit COPD teilnahmen, war die Anzahl der Studienteilnehmer mit Abweichungen vom korrigierten QT-Intervall von 30 bis 60 ms zu Studienbeginn in der SPIRIVA 9 mcg HANDIHALER-Gruppe höher als im Vergleich mit Placebo. Dieser Unterschied war offensichtlich, wenn sowohl die Bazett (QTcB) [20 (20 %) Patienten vs. 12 (12 %) Patienten] als auch die Fredericia (QTcF) [16 (16 %) Patienten vs. 1 (1 %) Patient] Korrekturen von vorgenommen wurden QT für Herzfrequenz. Kein Patient in beiden Gruppen hatte entweder ein QTcB oder ein QTcF von > 500 ms. Andere klinische Studien mit SPIRIVA HANDIHALER zeigten keine Wirkung des Arzneimittels auf die QTc-Intervalle.
Die Wirkung von Tiotropium-Trockenpulver zur Inhalation auf das QT-Intervall wurde auch in einer randomisierten, placebo- und positiv-kontrollierten Crossover-Studie mit 53 gesunden Freiwilligen untersucht. Die Probanden erhielten Tiotropium-Trockenpulver zur Inhalation 18 µg, 54 µg (das Dreifache der empfohlenen Dosis) oder Placebo für 12 Tage. EKG-Bewertungen wurden zu Studienbeginn und während des gesamten Dosierungsintervalls nach der ersten und letzten Dosis der Studienmedikation durchgeführt. Im Vergleich zu Placebo betrug die maximale mittlere Veränderung des studienspezifischen QTc-Intervalls gegenüber dem Ausgangswert 3,2 ms bzw. 0,8 ms für Tiotropium-Trockenpulver zur Inhalation 18 µg bzw. 54 µg. Kein Patient zeigte einen neuen Beginn von QTc > 500 ms oder QTc-Veränderungen von ≥ 60 ms gegenüber dem Ausgangswert.
Pharmakokinetik
Tiotropium wird durch Trockenpulverinhalation verabreicht. Einige der unten beschriebenen pharmakokinetischen Daten wurden mit höheren Dosen als für die Therapie empfohlen erhalten. Eine spezielle pharmakokinetische Studie bei Patienten mit COPD, in der einmal täglich Tiotropium aus dem RESPIMAT-Inhalator (5 µg) und als Inhalationspulver (18 µg) aus dem HANDIHALER-Gerät untersucht wurde, ergab eine ähnliche systemische Exposition zwischen den beiden Produkten.
Absorption
Nach Inhalation des Trockenpulvers durch junge gesunde Freiwillige deutet die absolute Bioverfügbarkeit von 19,5 % darauf hin, dass die Fraktion, die die Lunge erreicht, hochgradig bioverfügbar ist. Orale Lösungen von Tiotropium haben eine absolute Bioverfügbarkeit von 2-3 %. Es ist nicht zu erwarten, dass Nahrung die Resorption von Tiotropium beeinflusst. Maximale Tiotropium-Plasmakonzentrationen wurden 7 Minuten nach der Inhalation beobachtet.
Verteilung
Tiotropium wird zu 72 % an Plasmaproteine gebunden und hatte nach intravenöser Verabreichung an junge gesunde Probanden ein Verteilungsvolumen von 32 l/kg. Lokale Konzentrationen in der Lunge sind nicht bekannt, aber die Art der Verabreichung lässt auf wesentlich höhere Konzentrationen in der Lunge schließen. Studien an Ratten haben gezeigt, dass Tiotropium die Blut-Hirn-Schranke nicht leicht durchdringt.
Beseitigung
Die terminale Halbwertszeit von Tiotropium bei COPD-Patienten nach einmal täglicher Inhalation von 5 µg Tiotropium betrug etwa 25 Stunden. Die Gesamtclearance betrug nach intravenöser Verabreichung bei jungen gesunden Probanden 880 ml/min. Nach chronischer einmal täglicher Trockenpulverinhalation bei COPD-Patienten wurde der pharmakokinetische Steady State am Tag 7 erreicht, ohne dass es danach zu einer Akkumulation kam.
Stoffwechsel
Der Umfang des Stoffwechsels ist gering. Dies geht aus einer Urinausscheidung von 74 % unveränderter Substanz nach intravenöser Gabe an junge gesunde Probanden hervor. Tiotropium, ein Ester, wird nichtenzymatisch in den Alkohol N-Methylscopin und Dithienylglykolsäure gespalten, die beide nicht an Muskarinrezeptoren binden.
In-vitro-Experimente mit menschlichen Lebermikrosomen und menschlichen Hepatozyten deuten darauf hin, dass ein Bruchteil der verabreichten Dosis (74 % einer intravenösen Dosis werden unverändert im Urin ausgeschieden, 25 % verbleiben für den Metabolismus) durch Cytochrom-P450-abhängige Oxidation und anschließende Glutathion-Konjugation metabolisiert wird zu einer Vielzahl von Phase-II-Metaboliten. Dieser enzymatische Weg kann durch CYP450 2D6- und 3A4-Inhibitoren wie Chinidin, Ketoconazol und Gestoden gehemmt werden. Somit sind CYP450 2D6 und 3A4 an dem Stoffwechselweg beteiligt, der für die Elimination eines kleinen Teils der verabreichten Dosis verantwortlich ist. In-vitro-Studien mit menschlichen Lebermikrosomen zeigten, dass Tiotropium in supratherapeutischen Konzentrationen CYP450 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 oder 3A4 nicht hemmte.
Ausscheidung
Intravenös verabreichtes Tiotropiumbromid wird hauptsächlich unverändert im Urin ausgeschieden (74 %). Nach Inhalation von Trockenpulver bei COPD-Patienten im Steady State betrug die Urinausscheidung 7 % (1,3 Mikrogramm) der unveränderten Dosis über 24 Stunden. Die renale Clearance von Tiotropium übersteigt die Kreatinin-Clearance, was auf eine Ausscheidung in den Urin hindeutet.
Spezifische Populationen
Geriatrische Patienten
Wie für alle überwiegend renal ausgeschiedenen Arzneimittel zu erwarten, war mit fortschreitendem Alter eine Abnahme der renalen Clearance von Tiotropium verbunden (365 ml/min bei COPD-Patienten
Nierenfunktionsstörung
Nach 4-wöchiger Gabe von SPIRIVA 9 µg HANDIHALER oder SPIRIVA 9 µg RESPIMAT einmal täglich bei Patienten mit COPD führte eine leichte Nierenfunktionsstörung (Kreatinin-Clearance 60-
Leberfunktionsstörung
Die Auswirkungen einer Leberfunktionsstörung auf die Pharmakokinetik von Tiotropium wurden nicht untersucht.
Wechselwirkungen mit anderen Medikamenten
Es wurde eine Wechselwirkungsstudie mit Tiotropium (14,4 µg intravenöse Infusion über 15 Minuten) und Cimetidin 400 mg dreimal täglich oder Ranitidin 300 mg einmal täglich durchgeführt. Die gleichzeitige Gabe von Cimetidin und Tiotropium führte zu einem Anstieg der AUC0-4h um 20 %, einer Abnahme der renalen Clearance von Tiotropium um 28 % und keiner signifikanten Veränderung der Cmax und der über 96 Stunden im Urin ausgeschiedenen Menge. Die gleichzeitige Anwendung von Tiotropium mit Ranitidin hatte keinen Einfluss auf die Pharmakokinetik von Tiotropium.
Häufige Begleitmedikationen (langwirksame beta2-adrenerge Agonisten (LABA), inhalative Kortikosteroide (ICS)), die von Patienten mit COPD angewendet wurden, veränderten die Tiotropium-Exposition nicht.
Klinische Studien
Das klinische Entwicklungsprogramm von SPIRIVA 9 µg HANDIHALER (Tiotropiumbromid-Pulver zur Inhalation) bestand aus sechs Phase-3-Studien mit 2.663 Patienten mit COPD (1.308 erhielten SPIRIVA 9 µg HANDIHALER): zwei placebokontrollierte Studien über 1 Jahr, zwei placebokontrollierte Studien über 6 Monate Studien und zwei 1-jährige, Ipratropium-kontrollierte Studien. In diese Studien wurden Patienten aufgenommen, die eine klinische COPD-Diagnose hatten, 40 Jahre oder älter waren, in der Vorgeschichte mehr als 10 Packungsjahre geraucht hatten und ein forciertes Ausatmungsvolumen in einer Sekunde (FEV1) von weniger als oder gleich 60 % aufwiesen. oder 65 % des Sollwerts und ein Verhältnis von FEV1/FVC von kleiner oder gleich 0,7.
In diesen Studien bewirkte SPIRIVA HANDIHALER, einmal täglich morgens verabreicht, eine Verbesserung der Lungenfunktion (FEV1), wobei die maximale Wirkung innerhalb von 3 Stunden nach der ersten Dosis eintrat.
Zwei weitere Studien untersuchten Exazerbationen: eine 6-monatige, randomisierte, doppelblinde, placebokontrollierte, multizentrische klinische Studie mit 1829 COPD-Patienten in einer US-amerikanischen Veterans Affairs-Umgebung und eine 4-jährige, randomisierte, doppelblinde, placebokontrollierte, multizentrische, klinische Studie mit 5992 COPD-Patienten. Langzeitwirkungen auf die Lungenfunktion und andere Ergebnisse wurden ebenfalls in der 4-jährigen multizentrischen Studie bewertet.
Auswirkungen von 6 Monaten bis 1 Jahr auf die Lungenfunktion
In den placebokontrollierten 1-Jahres-Studien betrug die mittlere Verbesserung des FEV1 nach 30 Minuten 0,13 Liter (13 %) mit einer Spitzenverbesserung von 0,24 Liter (24 %) im Vergleich zum Ausgangswert nach der ersten Dosis (Tag 1). Weitere Verbesserungen des FEV1 und der forcierten Vitalkapazität (FVC) wurden mit einem pharmakodynamischen Steady State beobachtet, der bei einmal täglicher Behandlung an Tag 8 erreicht wurde. Die mittlere maximale Verbesserung des FEV1 im Vergleich zum Ausgangswert betrug 0,28 bis 0,31 Liter (28 % bis 31 %) nach 1 Woche (Tag 8) einmal täglicher Behandlung. Die Verbesserung der Lungenfunktion wurde nach einer Einzeldosis 24 Stunden lang aufrechterhalten und über den 1-jährigen Behandlungszeitraum durchgehend ohne Anzeichen einer Toleranz aufrechterhalten.
In den beiden 6-monatigen placebokontrollierten Studien wurden serielle spirometrische Auswertungen während der Tageszeit in Studie A (12 Stunden) durchgeführt und in Studie B auf 3 Stunden begrenzt. Die seriellen FEV1-Werte über 12 Stunden (Studie A) werden in angezeigt Abbildung 1. Diese Studien untermauern die Verbesserung der Lungenfunktion (FEV1) mit SPIRIVA 9 mcg HANDIHALER, die über den spirometrischen Beobachtungszeitraum anhielt. Die Wirksamkeit wurde für 24 Stunden nach der Verabreichung über den 6-monatigen Behandlungszeitraum aufrechterhalten.
Abbildung 1: Mittleres FEV1 im Zeitverlauf (vor und nach Verabreichung des Studienmedikaments) an den Tagen 1 und 169 für Studie A (eine sechsmonatige placebokontrollierte Studie)* 
*Mittelwerte, bereinigt um Zentrum, Behandlung und Basislinieneffekt. An Tag 169 schlossen insgesamt 183 bzw. 149 Patienten in der SPIRIVA HANDIHALER- bzw. Placebo-Gruppe die Studie ab. Die Daten für die verbleibenden Patienten wurden unter Verwendung der letzten Beobachtung oder der ungünstigsten Beobachtung, die fortgeschrieben wurde, imputiert.
Die Ergebnisse jeder der Ipratropium-kontrollierten 1-Jahres-Studien waren den Ergebnissen der 1-Jahres-Placebo-kontrollierten Studien ähnlich. Die Ergebnisse einer dieser Studien sind in Abbildung 2 dargestellt.
Abbildung 2: Mittleres FEV1 über die Zeit (0 bis 6 Stunden nach der Dosis) an den Tagen 1 und 92, jeweils für eine der beiden Ipratropium-kontrollierten Studien* 
*Mittelwerte, bereinigt um Zentrum, Behandlung und Basislinieneffekt. An Tag 92 (primärer Endpunkt) schlossen insgesamt 151 bzw. 69 Patienten in der SPIRIVA HANDIHALER- bzw. Ipratropium-Gruppe die 3-monatige Beobachtung ab. Die Daten für die verbleibenden Patienten wurden unter Verwendung der letzten Beobachtung oder der ungünstigsten Beobachtung, die fortgeschrieben wurde, imputiert.
Eine randomisierte, placebokontrollierte klinische Studie an 105 COPD-Patienten zeigte, dass die Bronchodilatation im Vergleich zu Placebo über das gesamte 24-stündige Dosierungsintervall aufrechterhalten wurde, unabhängig davon, ob SPIRIVA 9 µg HANDIHALER morgens oder abends verabreicht wurde.
In den beiden Placebo-kontrollierten Studien hatten die Patienten, die SPIRIVA 9 mcg HANDIHALER einnahmen, in jeder Woche des einjährigen Behandlungszeitraums einen geringeren Bedarf an kurz wirkenden Beta2-Agonisten zur Notfallbehandlung. In einer der beiden 6-Monats-Studien wurde im Vergleich zu Placebo eine Verringerung der Anwendung kurz wirksamer Beta2-Agonisten als Notfallbehandlung nachgewiesen.
4-Jahres-Auswirkungen auf die Lungenfunktion
Eine 4-jährige, randomisierte, doppelblinde, placebokontrollierte, multizentrische klinische Studie mit 5992 COPD-Patienten wurde durchgeführt, um die langfristigen Wirkungen von SPIRIVA HANDIHALER auf das Fortschreiten der Krankheit (Abnahmerate von FEV1) zu bewerten. Die Patienten durften alle Atemwegsmedikamente (einschließlich kurz- und langwirksamer Beta-Agonisten, inhalative und systemische Steroide und Theophylline) mit Ausnahme von inhalativen Anticholinergika verwenden. Die Patienten waren 40 bis 88 Jahre alt, 75 % männlich und zu 90 % Kaukasier mit einer COPD-Diagnose und einem mittleren FEV1 vor Bronchodilatation von 39 % (Bereich = 9 % bis 76 %) bei Studieneintritt. Es gab keinen Unterschied zwischen den Gruppen bei einem der co-primären Wirksamkeitsendpunkte, der jährlichen Abnahmerate des FEV1 vor und nach der Bronchodilatation, wie durch ähnliche Steigungen der FEV1-Abnahme im Laufe der Zeit gezeigt wurde (Abbildung 3).
SPIRIVA HANDIHALER hielt die Verbesserungen des FEV1-Talwerts (vor der Verabreichung) (angepasster Mittelwert im Laufe der Zeit: 87 bis 103 ml) während der 4 Jahre der Studie aufrecht (Abbildung 3).
Abbildung 3: FEV1-Mittelwerte (vor der Dosis) zu jedem Zeitpunkt 
ANOVA mit wiederholter Messung wurde verwendet, um Mittelwerte abzuschätzen. Die Mittelwerte werden für Basislinienmessungen angepasst. Baseline-Tal-FEV1 (beobachteter Mittelwert) = 1,12. Patienten mit ≥3 akzeptablen Lungenfunktionstests nach Tag 30 und einem nicht fehlenden Ausgangswert wurden in die Analyse eingeschlossen.
Exazerbationen
Die Wirkung von SPIRIVA 9 mcg HANDIHALER auf COPD-Exazerbationen wurde in zwei klinischen Studien untersucht: einer oben beschriebenen 4-jährigen klinischen Studie und einer 6-monatigen klinischen Studie mit 1829 COPD-Patienten in einer Veterans Affairs-Umgebung. In der 6-monatigen Studie wurden COPD-Exazerbationen als ein Komplex von respiratorischen Symptomen (Zunahme oder Neuauftreten) von mehr als einem der folgenden definiert: Husten, Auswurf, Keuchen, Dyspnoe oder Engegefühl in der Brust mit einer Dauer von mindestens 3 Tagen die eine Behandlung mit Antibiotika, systemischen Steroiden oder einen Krankenhausaufenthalt erfordern. Die Bevölkerung war zwischen 40 und 90 Jahre alt, wobei 99 % Männer, 91 % Kaukasier waren, und hatte COPD mit einem erwarteten mittleren FEV1-Prozentsatz vor Bronchodilatation von 36 % (Bereich = 8 % bis 93 %). Die Patienten durften neben inhalativen Anticholinergika auch Atemwegsmedikamente (einschließlich kurz- und langwirksamer Beta-Agonisten, inhalative und systemische Steroide und Theophylline) anwenden. In der 6-monatigen Studie waren die co-primären Endpunkte der Anteil der Patienten mit einer COPD-Exazerbation und der Anteil der Patienten mit Krankenhausaufenthalt aufgrund einer COPD-Exazerbation. SPIRIVA 9 mcg HANDIHALER reduzierte den Anteil der COPD-Patienten, bei denen Exazerbationen auftraten, im Vergleich zu Placebo signifikant (27,9 % vs. 32,3 %; Odds Ratio (OR) (Tiotropium/Placebo) = 0,81; 95 %-KI = 0,66; 0,99; p = 0,037). ). Der Anteil der Patienten, die aufgrund von COPD-Exazerbationen ins Krankenhaus eingeliefert wurden, betrug bei Patienten, die SPIRIVA 9 mcg HANDIHALER verwendeten, im Vergleich zu Placebo 7,0 % vs. 9,5 %; ODER = 0,72; 95 % KI = 0,51, 1,01; p = 0,056.
Exazerbationen wurden als sekundärer Endpunkt in der 4-jährigen multizentrischen Studie ausgewertet. In dieser Studie wurden COPD-Exazerbationen definiert als eine Zunahme oder ein erneutes Auftreten von mehr als einem der folgenden Atemwegssymptome (Husten, Auswurf, Auswurfeiterung, Keuchen, Atemnot) mit einer Dauer von drei oder mehr Tagen, die eine Behandlung mit Antibiotika und/oder erfordern systemische (orale, intramuskuläre oder intravenöse) Steroide. SPIRIVA HANDIHALER reduzierte signifikant das Risiko einer Exazerbation um 14 % (Hazard Ratio (HR) = 0,86; 95 % KI = 0,81, 0,91; p
Gesamtmortalität
In der oben beschriebenen 4-jährigen placebokontrollierten Lungenfunktionsstudie wurde die Gesamtmortalität im Vergleich zu Placebo bewertet. Es gab keine signifikanten Unterschiede in der Gesamtsterblichkeitsrate zwischen SPIRIVA 9 mcg HANDIHALER und Placebo.
Die Gesamtmortalität von SPIRIVA HANDIHALER wurde auch mit Tiotropium-Inhalationsspray 5 µg (SPIRIVA RESPIMAT 5 µg) in einer zusätzlichen randomisierten, doppelblinden, aktiv kontrollierten Double-Dummy-Studie mit einem Beobachtungszeitraum von bis zu 3 Jahren verglichen. Die Gesamtmortalität war bei SPIRIVA 9 mcg HANDIHALER und SPIRIVA RESPIMAT ähnlich.
INFORMATIONEN ZUM PATIENTEN
SPIRIVA 9mcgR® (speh REE vah) HANDIHALER® (Tiotropiumbromid) Pulver zur Inhalation zur oralen Inhalation
SPIRIVA-Kapseln NICHT schlucken.
Wichtige Informationen: SPIRIVA-Kapseln nicht schlucken. SPIRIVA 9 mcg-Kapseln sollten nur mit dem HANDIHALER-Gerät verwendet und durch den Mund inhaliert werden (orale Inhalation).
Lesen Sie die Informationen, die Ihrem SPIRIVA HANDIHALER beiliegen, bevor Sie ihn verwenden und jedes Mal, wenn Sie Ihr Rezept nachfüllen. Möglicherweise gibt es neue Informationen. Diese Packungsbeilage ersetzt nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung.
Was ist SPIRIVA HANDIHALER?
SPIRIVA 9mcg HANDIHALER ist kein Notfallarzneimittel und sollte nicht zur Behandlung von plötzlichen Atemproblemen verwendet werden. Ihr Arzt kann Ihnen andere Arzneimittel zur Anwendung bei plötzlichen Atemproblemen verschreiben.
Es ist nicht bekannt, ob SPIRIVA 9mcg HANDIHALER bei Kindern sicher und wirksam ist.
Wer sollte SPIRIVA 9mcg HANDIHALER nicht einnehmen?
Verwenden Sie SPIRIVA 9mcg HANDIHALER nicht, wenn Sie:
Zu den Symptomen einer schweren allergischen Reaktion auf SPIRIVA 9mcg HANDIHALER können gehören:
Wenn Sie diese Symptome einer allergischen Reaktion haben, beenden Sie die Einnahme von SPIRIVA HANDIHALER und rufen Sie sofort Ihren Arzt an oder suchen Sie die Notaufnahme des nächstgelegenen Krankenhauses auf.
Was sollte ich meinem Arzt vor der Anwendung von SPIRIVA HANDIHALER mitteilen?
Informieren Sie Ihren Arzt vor der Einnahme von SPIRIVA HANDIHALER über alle Ihre Erkrankungen, auch wenn Sie:
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und nicht verschreibungspflichtiger Arzneimittel und Augentropfen, Vitamine und Kräuterergänzungen. Einige Ihrer anderen Arzneimittel oder Nahrungsergänzungsmittel können die Wirkungsweise von SPIRIVA HANDIHALER beeinflussen. SPIRIVA 9mcg HANDIHALER ist ein Anticholinergikum. Sie sollten während der Anwendung von SPIRIVA 9mcg HANDIHALER keine anderen Anticholinergika einnehmen, einschließlich Ipratropium. Fragen Sie Ihren Arzt oder Apotheker, wenn Sie sich nicht sicher sind, ob eines Ihrer Arzneimittel ein Anticholinergikum ist.
Informieren Sie sich über die Medikamente, die Sie einnehmen. Führen Sie eine Liste Ihrer Arzneimittel mit sich, damit Sie sie Ihrem Arzt und Apotheker zeigen können, wenn Sie ein neues Arzneimittel erhalten.
Wie sollte ich SPIRIVA 9mcg HANDIHALER einnehmen?
Was sollte ich bei der Anwendung von SPIRIVA 9mcg HANDIHALER vermeiden?
Welche Nebenwirkungen kann SPIRIVA HANDIHALER haben?
SPIRIVA 9mcg HANDIHALER kann schwerwiegende Nebenwirkungen hervorrufen, darunter: Allergische Reaktionen. Zu den Symptomen können gehören:
Wenn Sie diese Symptome einer allergischen Reaktion haben, beenden Sie die Einnahme von SPIRIVA HANDIHALER und rufen Sie sofort Ihren Arzt an oder suchen Sie die Notaufnahme des nächstgelegenen Krankenhauses auf.
Wenn Sie diese Symptome eines Bronchospasmus haben, beenden Sie die Einnahme von SPIRIVA 9 mcg HANDIHALER und rufen Sie sofort Ihren Arzt an oder suchen Sie die Notaufnahme des nächstgelegenen Krankenhauses auf.
Die alleinige Anwendung von Augentropfen zur Behandlung dieser Symptome funktioniert möglicherweise nicht. Wenn Sie diese Symptome haben, beenden Sie die Einnahme von SPIRIVA HANDIHALER und rufen Sie sofort Ihren Arzt an.
Wenn Sie diese Symptome einer Harnverhaltung haben, beenden Sie die Einnahme von SPIRIVA HANDIHALER und wenden Sie sich sofort an Ihren Arzt.
Andere Nebenwirkungen von SPIRIVA 9mcg HANDIHALER umfassen:
Dies sind nicht alle möglichen Nebenwirkungen von SPIRIVA HANDIHALER. Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist SPIRIVA HANDIHALER aufzubewahren?
Fragen Sie Ihren Arzt oder Apotheker, wenn Sie Fragen zur Aufbewahrung Ihrer SPIRIVA 9mcg-Kapseln haben.
Bewahren Sie SPIRIVA HANDIHALER, SPIRIVA 9-mcg-Kapseln und alle Arzneimittel außerhalb der Reichweite von Kindern auf.
Allgemeine Informationen über SPIRIVA HANDIHALER
Arzneimittel werden manchmal für andere als die in den Packungsbeilagen aufgeführten Zwecke verschrieben. Verwenden Sie SPIRIVA 9mcg HANDIHALER nicht für einen Zweck, für den es nicht verschrieben wurde. Geben Sie SPIRIVA HANDIHALER nicht an andere Personen weiter, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Für weitere Informationen über SPIRIVA HANDIHALER sprechen Sie mit Ihrem Arzt. Sie können Ihren Arzt oder Apotheker um Informationen über SPIRIVA 9mcg HANDIHALER bitten, die für Angehörige der Gesundheitsberufe bestimmt sind.
Weitere Informationen über SPIRIVA 9mcg HANDIHALER erhalten Sie unter www.SPIRIVA.com, oder scannen Sie den Code unten oder rufen Sie Boehringer Ingelheim Pharmaceuticals, Inc. unter 1-800-542-6257 oder (TTY) 1-800-459-9906 an.
Was sind die Inhaltsstoffe von SPIRIVA 9mcg HANDIHALER?
Wirkstoff: Tiotropium
Inaktiver Inhaltsstoff: Lactose-Monohydrat
Was ist COPD (chronisch obstruktive Lungenerkrankung)?
COPD ist eine schwere Lungenerkrankung, die chronische Bronchitis, Emphysem oder beides umfasst. Die meisten COPD werden durch Rauchen verursacht. Wenn Sie COPD haben, verengen sich Ihre Atemwege. Die Luft strömt also langsamer aus Ihren Lungen. Dadurch wird das Atmen erschwert.
Diese Patienteninformation wurde von der US Food and Drug Administration genehmigt.
GEBRAUCHSANWEISUNGSPIRIVA® (speh REE vah) HANDIHALER® (Tiotropiumbromid) Pulver zur Inhalation zur oralen Inhalation
SPIRIVA-Kapseln nicht schlucken.
Wichtige Informationen zur Verwendung Ihres SPIRIVA HANDIHALER
Lesen Sie zuerst die Patienteninformationen und dann diese Gebrauchsanweisung, bevor Sie mit der Anwendung von SPIRIVA HANDIHALER beginnen und jedes Mal, wenn Sie Ihr Rezept nachfüllen. Möglicherweise gibt es neue Informationen.
Machen Sie sich mit Ihrem HANDIHALER-Gerät und den SPIRIVA-Kapseln vertraut:
Ihr SPIRIVA HANDIHALER wird mit SPIRIVA 9 mcg-Kapseln in Blisterverpackung und einem HANDIHALER-Gerät geliefert. Verwenden Sie das mit Ihrem Arzneimittel gelieferte neue HANDIHALER-Gerät.
Zu den Teilen Ihres HANDIHALER-Geräts gehören:
(Siehe Abbildung A)
Abbildung A
Jede SPIRIVA 9mcg-Kapsel ist in einer Blisterpackung verpackt. (Siehe Abbildung B)
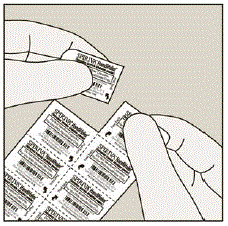
Abbildung B

Abbildung C
Die Einnahme Ihrer vollständigen Tagesdosis des Arzneimittels erfordert 4 Hauptschritte.
Schritt 1. Öffnen Ihres HANDIHALER-Geräts:
Nachdem Sie Ihr HANDIHALER-Gerät aus dem Beutel genommen haben:
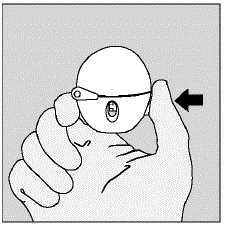
Abbildung D
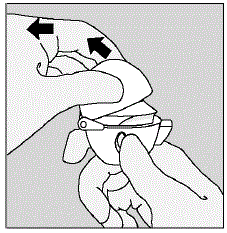
Abbildung E

Abbildung F
Schritt 2. Einsetzen der SPIRIVA 9mcg-Kapsel in Ihr HANDIHALER-Gerät:
Trennen Sie jeden Tag nur 1 Blister von der Blisterkarte, indem Sie ihn entlang der perforierten Linie abreißen. (Siehe Abbildung G)
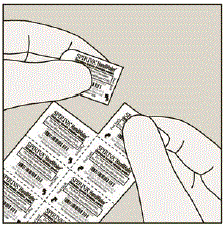
Abbildung G
Entfernen Sie die SPIRIVA-Kapsel aus der Blisterpackung:
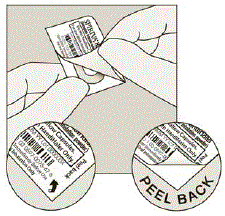
Abbildung H
Legen Sie die SPIRIVA-9-mcg-Kapsel in die mittlere Kammer Ihres HANDIHALER-Geräts. (Siehe Abbildung I)
Abbildung I
Schließen Sie das Mundstück fest gegen die graue Basis, bis Sie ein Klicken hören. Lassen Sie die Staubkappe (Deckel) geöffnet. (Siehe Abbildung J)
Abbildung J
Schritt 3. Durchstechen der SPIRIVA 9mcg-Kapsel:
Abbildung K
Schritt 4. Einnahme Ihrer vollen Tagesdosis (2 Inhalationen aus derselben SPIRIVA 9-mcg-Kapsel):
Atmen Sie in einem Atemzug vollständig aus, Entleeren Sie Ihre Lungen von jeglicher Luft. (Siehe Abbildung L)
Abbildung L
Wichtig: Nicht Atmen Sie in Ihr HANDIHALER-Gerät ein.
Nehmen Sie mit Ihrem nächsten Atemzug Ihre Medizin ein:
Abbildung M
Die Rassel signalisiert Ihnen, dass Sie richtig eingeatmet haben. Wenn Sie kein Rasseln hören oder fühlen, lesen Sie den Abschnitt „Wenn Sie beim Einatmen Ihres Arzneimittels kein Rasseln der SPIRIVA-Kapsel hören oder fühlen“.
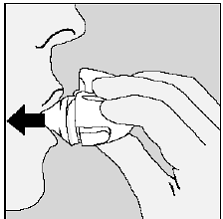
Abbildung N
Um Ihre volle Tagesdosis zu erhalten, müssen Sie aus derselben SPIRIVA-Kapsel erneut vollständig ausatmen (siehe Abbildung N) und ein zweites Mal einatmen (siehe Abbildung O).
Wichtig: Nicht Drücken Sie erneut die grüne Durchstechtaste.
Abbildung O
Denken Sie daran: Um Ihre volle Arzneimitteldosis jeden Tag zu erhalten, müssen Sie zweimal aus derselben SPIRIVA 9-mcg-Kapsel einatmen. Achten Sie darauf, dass Sie jedes Mal vollständig ausatmen, bevor Sie mit Ihrem HANDIHALER-Gerät einatmen.
Pflege und Aufbewahrung Ihres SPIRIVA HANDIHALER:
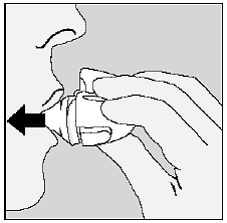
Abbildung P
Wenn Sie die SPIRIVA 9mcg-Kapsel beim Einatmen Ihres Arzneimittels nicht hören oder fühlen:
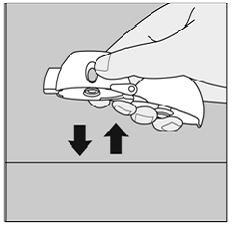
Abbildung Q
Unterlassen Sie Drücken Sie erneut die grüne Durchstechtaste.
Halten Sie Ihr HANDIHALER-Gerät mit dem Mundstück nach oben und klopfen Sie Ihr HANDIHALER-Gerät sanft auf einen Tisch. (Siehe Abbildung Q)
Überprüfen Sie, ob das Mundstück vollständig geschlossen ist. Atmen Sie vollständig aus, bevor Sie mit dem Mundstück im Mund wieder tief einatmen. (Siehe Abbildung O)
Wenn Sie nach Wiederholung der obigen Schritte immer noch kein Rasseln der SPIRIVA-Kapsel hören oder fühlen:
Reinigung Ihres HANDIHALER-Geräts:
Reinigen Sie Ihr HANDIHALER-Gerät nach Bedarf. (Siehe Abbildung R)
Reinigungsschritte:
Abbildung R
Hilfreiche Tipps, um sicherzustellen, dass Sie Ihre volle Tagesdosis SPIRIVA 9 mcg HANDIHALER richtig einnehmen:
Wenden Sie sich für weitere Informationen an Ihren Arzt oder Apotheker oder gehen Sie zu www.spiriva.com, scannen Sie den Code unten oder rufen Sie 1-800-542-6257 oder (TTY) 1-800-459-9906 an.
Diese Gebrauchsanweisung wurde von der US Food and Drug Administration genehmigt.
Was ist Strattera 40 mg und wie wird es angewendet?
Strattera ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome der Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS). Strattera 18 mg kann allein oder mit anderen Medikamenten verwendet werden.
Strattera 10 mg ist ein selektiver Noradrenalin-Wiederaufnahmehemmer.
Es ist nicht bekannt, ob Strattera bei Kindern unter 2 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Strattera haben?
Strattera 25 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Strattera 40 mg gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Strattera. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
WARNUNG
SELBSTMORDGEDANKEN BEI KINDERN UND JUGENDLICHEN
STRATTERA (Atomoxetin) erhöhte das Risiko von Suizidgedanken in Kurzzeitstudien bei Kindern oder Jugendlichen mit Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS). Jeder, der die Anwendung von STRATTERA 18 mg bei einem Kind oder Jugendlichen in Betracht zieht, muss dieses Risiko mit der klinischen Notwendigkeit abwägen. Komorbiditäten, die bei ADHS auftreten, können mit einem erhöhten Risiko für Suizidgedanken und/oder -verhalten verbunden sein. Patienten, die mit einer Therapie begonnen werden, sollten engmaschig auf Suizidalität (Suizidgedanken und -verhalten), klinische Verschlechterung oder ungewöhnliche Verhaltensänderungen überwacht werden. Familien und Betreuer sollten auf die Notwendigkeit einer genauen Beobachtung und Kommunikation mit dem verschreibenden Arzt hingewiesen werden. STRATTERA 10 mg ist für ADHS bei pädiatrischen und erwachsenen Patienten zugelassen. STRATTERA 40 mg ist nicht für schwere depressive Störungen zugelassen. Gepoolte Analysen von Placebo-kontrollierten Kurzzeitstudien (6 bis 18 Wochen) mit STRATTERA 10 mg bei Kindern und Jugendlichen (insgesamt 12 Studien mit über 2200 Patienten, darunter 11 Studien zu ADHS und 1 Studie zu Enuresis) haben ein größeres Risiko ergeben Suizidgedanken früh während der Behandlung bei Patienten, die STRATTERA erhielten, im Vergleich zu Placebo. Das durchschnittliche Risiko für Suizidgedanken bei Patienten, die STRATTERA 25 mg erhielten, betrug 0,4 % (5/1357 Patienten), verglichen mit keinem Risiko bei den mit Placebo behandelten Patienten (851 Patienten). In diesen Studien kam es zu keinen Suiziden [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
BEZEICHNUNG
STRATTERA® (Atomoxetin) ist ein selektiver Noradrenalin-Wiederaufnahmehemmer. Atomoxetin-HCl ist das R(-)-Isomer, wie durch Röntgenbeugung bestimmt. Die chemische Bezeichnung lautet (-)-N-Methyl-3-phenyl-3-(o-tolyloxy)-propylaminhydrochlorid. Die Summenformel lautet C17H21NO•HCl, was einem Molekulargewicht von 291,82 entspricht. Die chemische Struktur ist:
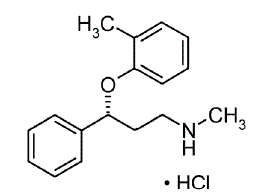
Atomoxetin-HCl ist ein weißer bis praktisch weißer Feststoff mit einer Löslichkeit von 27,8 mg/ml in Wasser.
STRATTERA-Kapseln sind nur zur oralen Verabreichung bestimmt.
Jede Kapsel enthält Atomoxetin-HCl entsprechend 10, 18, 25, 40, 60, 80 oder 100 mg Atomoxetin. Die Kapseln enthalten außerdem Quellstärke und Dimethicon. Die Kapselhüllen enthalten Gelatine, Natriumlaurylsulfat und andere inaktive Inhaltsstoffe. Die Kapselhüllen enthalten außerdem einen oder mehrere der folgenden Stoffe:
FD&C Blau Nr. 2, synthetisches gelbes Eisenoxid, Titandioxid, rotes Eisenoxid. Die Kapseln sind mit essbarer schwarzer Tinte bedruckt.
INDIKATIONEN
Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS)
STRATTERA 25 mg ist angezeigt zur Behandlung von Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS).
Die Wirksamkeit von STRATTERA-Kapseln wurde in sieben klinischen Studien bei ambulanten Patienten mit ADHS nachgewiesen: vier 6- bis 9-wöchige Studien mit pädiatrischen Patienten (im Alter von 6 bis 18 Jahren), zwei 10-wöchige Studien mit Erwachsenen und eine Erhaltungsstudie in der Pädiatrie (im Alter von 6 zu 15) [vgl Klinische Studien ].
Diagnostische Überlegungen
Eine Diagnose von ADHS (DSM-IV) impliziert das Vorhandensein von hyperaktiv-impulsiven oder unaufmerksamen Symptomen, die eine Beeinträchtigung verursachen und vor dem Alter von 7 Jahren vorhanden waren. Die Symptome müssen anhaltend sein, schwerer sein als typischerweise bei Personen auf einem vergleichbaren Entwicklungsstand beobachtet, müssen eine klinisch signifikante Beeinträchtigung verursachen, z. zB Schule (oder Arbeit) und zu Hause. Die Symptome dürfen nicht besser durch eine andere psychische Störung erklärt werden.
Die spezifische Ätiologie von ADHS ist unbekannt, und es gibt keinen einzigen diagnostischen Test. Eine adäquate Diagnostik erfordert nicht nur den Einsatz medizinischer, sondern auch spezieller psychologischer, pädagogischer und sozialer Ressourcen. Das Lernen kann beeinträchtigt sein oder auch nicht. Die Diagnose muss auf einer vollständigen Anamnese und Beurteilung des Patienten beruhen und nicht nur auf dem Vorhandensein der erforderlichen Anzahl von DSM-IV-Merkmalen.
Beim unaufmerksamen Typ müssen mindestens 6 der folgenden Symptome seit mindestens 6 Monaten bestehen: Mangel an Aufmerksamkeit für Details/Falschfehler, Mangel an nachhaltiger Aufmerksamkeit, schlechtes Zuhören, Nichtbefolgen von Aufgaben, schlechte Organisation, vermeidet Aufgaben erfordert anhaltende geistige Anstrengung, verliert Dinge, ist leicht ablenkbar, vergesslich. Beim hyperaktiv-impulsiven Typ müssen mindestens 6 der folgenden Symptome seit mindestens 6 Monaten bestehen: Zappeln/Winden, Verlassen des Sitzes, unangemessenes Laufen/Klettern, Schwierigkeiten bei leisen Aktivitäten, „unterwegs“, übermäßiges Reden, Platzen antwortet, kann es kaum erwarten, an der Reihe zu sein, aufdringlich. Für eine Diagnose des kombinierten Typs müssen sowohl die Kriterien für Unaufmerksamkeit als auch für Hyperaktivität und Impulsivität erfüllt sein.
Notwendigkeit eines umfassenden Behandlungsprogramms
STRATTERA 18 mg ist als integraler Bestandteil eines Gesamtbehandlungsprogramms für ADHS indiziert, das andere (psychologische, erzieherische, soziale) Maßnahmen für Patienten mit diesem Syndrom umfassen kann. Eine medikamentöse Behandlung ist möglicherweise nicht für alle Patienten mit diesem Syndrom indiziert. Die medikamentöse Behandlung ist nicht für die Anwendung bei Patienten bestimmt, die Symptome aufweisen, die auf Umweltfaktoren und/oder andere primäre psychiatrische Störungen, einschließlich Psychosen, zurückzuführen sind. Eine angemessene schulische Unterbringung ist bei Kindern und Jugendlichen mit dieser Diagnose unerlässlich, und psychosoziale Interventionen sind oft hilfreich. Wenn Heilmaßnahmen allein nicht ausreichen, hängt die Entscheidung über die Verschreibung von Medikamenten zur medikamentösen Behandlung von der Einschätzung des Arztes über die Chronizität und Schwere der Symptome des Patienten ab.
DOSIERUNG UND ANWENDUNG
Akute Behandlung
Dosierung von Kindern und Jugendlichen bis 70 kg Körpergewicht
STRATTERA 40 mg sollte mit einer täglichen Gesamtdosis von etwa 0,5 mg/kg begonnen und nach mindestens 3 Tagen auf eine angestrebte tägliche Gesamtdosis von etwa 1,2 mg/kg erhöht werden, verabreicht entweder als einzelne Tagesdosis am Morgen oder gleichmäßig verteilt Dosen morgens und am späten Nachmittag/frühen Abend. Für Dosen über 1,2 mg/kg/Tag wurde kein zusätzlicher Nutzen nachgewiesen [siehe Klinische Studien ].
Die tägliche Gesamtdosis bei Kindern und Jugendlichen sollte 1,4 mg/kg oder 100 mg nicht überschreiten, je nachdem, welcher Wert geringer ist.
Dosierung von Kindern und Jugendlichen über 70 kg Körpergewicht und Erwachsenen
STRATTERA 18 mg sollte mit einer täglichen Gesamtdosis von 40 mg begonnen und nach mindestens 3 Tagen auf eine angestrebte tägliche Gesamtdosis von etwa 80 mg erhöht werden, die entweder als einzelne Tagesdosis morgens oder in gleichmäßig verteilten Dosen morgens und morgens verabreicht wird später Nachmittag/früher Abend. Nach 2 bis 4 weiteren Wochen kann die Dosis bei Patienten, die kein optimales Ansprechen erreicht haben, auf maximal 100 mg erhöht werden. Es gibt keine Daten, die eine erhöhte Wirksamkeit bei höheren Dosen unterstützen [siehe Klinische Studien ].
Die maximal empfohlene Tagesgesamtdosis bei Kindern und Jugendlichen über 70 kg und Erwachsenen beträgt 100 mg.
Wartung/Erweiterte Behandlung
Es besteht allgemein Einigkeit darüber, dass eine pharmakologische Behandlung von ADHS über einen längeren Zeitraum erforderlich sein kann. In einer kontrollierten Studie wurde der Nutzen einer Erhaltungstherapie mit STRATTERA 18 mg bei pädiatrischen Patienten (Alter 6-15 Jahre) mit ADHS nach Erreichen eines Ansprechens in einem Dosisbereich von 1,2 bis 1,8 mg/kg/Tag nachgewiesen. Patienten, die STRATTERA in der Erhaltungsphase zugewiesen wurden, erhielten im Allgemeinen dieselbe Dosis, die verwendet wurde, um in der unverblindeten Phase ein Ansprechen zu erzielen. Der Arzt, der sich für die Anwendung von STRATTERA über einen längeren Zeitraum entscheidet, sollte den langfristigen Nutzen des Arzneimittels für den einzelnen Patienten regelmäßig neu bewerten [siehe Klinische Studien ].
Allgemeine Dosierungsinformationen
STRATTERA kann mit oder ohne Nahrung eingenommen werden.
STRATTERA 40 mg kann ohne Ausschleichen abgesetzt werden.
STRATTERA-Kapseln sind nicht zum Öffnen bestimmt, sie sollten unzerkaut eingenommen werden [siehe INFORMATIONEN ZUM PATIENTEN ].
Die Sicherheit von Einzeldosen über 120 mg und Tagesgesamtdosen über 150 mg wurde nicht systematisch untersucht.
Dosierung in bestimmten Populationen
Dosisanpassung für Patienten mit eingeschränkter Leberfunktion
Für ADHS-Patienten mit Leberinsuffizienz (HI) wird eine Dosisanpassung wie folgt empfohlen: Bei Patienten mit mittelschwerer HI (Child-Pugh-Klasse B) sollten Anfangs- und Zieldosis auf 50 % der normalen Dosis reduziert werden (bei Patienten ohne HALLO). Bei Patienten mit schwerer HI (Child-Pugh-Klasse C) sollten Anfangsdosis und Zieldosis auf 25 % des Normalwertes reduziert werden [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Dosisanpassung zur Anwendung mit einem starken CYP2D6-Inhibitor oder bei Patienten, die bekanntermaßen CYP2D6-PMs sind
Bei Kindern und Jugendlichen mit einem Körpergewicht bis zu 70 kg, denen starke CYP2D6-Inhibitoren, z. B. Paroxetin, Fluoxetin und Chinidin, verabreicht werden, oder bei Patienten, bei denen bekannt ist, dass sie CYP2D6-PMs sind, sollte STRATTERA mit 0,5 mg/kg/Tag begonnen und nur auf diese Dosis erhöht werden die übliche Zieldosis von 1,2 mg/kg/Tag, wenn sich die Symptome nach 4 Wochen nicht bessern und die Anfangsdosis gut vertragen wird.
Bei Kindern und Jugendlichen über 70 kg Körpergewicht und Erwachsenen, denen starke CYP2D6-Inhibitoren, z. B. Paroxetin, Fluoxetin und Chinidin, verabreicht werden, sollte STRATTERA mit 40 mg/Tag begonnen und nur dann auf die übliche Zieldosis von 80 mg/Tag erhöht werden, wenn die Symptome ausbleiben Besserung nach 4 Wochen und die Anfangsdosis wird gut vertragen.
WIE GELIEFERT
Darreichungsformen und Stärken
Jede Kapsel enthält Atomoxetin-HCl entsprechend 10 mg (Opaque White, Opaque White), 18 mg (Gold, Opaque White), 25 mg (Opaque Blue, Opaque White), 40 mg (Opaque Blue, Opaque Blue), 60 mg (Opaque Blau, Gold), 80 mg (Opaque Brown, Opaque White) oder 100 mg (Opaque Brown, Opaque Brown) Atomoxetin.
Lagerung und Handhabung
Bei 25 °C (77 °F) lagern; Abweichungen bis 15° bis 30°C (59° bis 86°F) zulässig [siehe USP Controlled Room Temperature].
Vertrieben durch: Lilly USA, LLC Indianapolis, IN 46285, USA. Überarbeitet: Mai 2017.
NEBENWIRKUNGEN
Erfahrung mit klinischen Studien
STRATTERA 10 mg wurde in klinischen Studien 5382 Kindern oder Jugendlichen mit ADHS und 1007 Erwachsenen mit ADHS verabreicht. Während der klinischen ADHS-Studien wurden 1625 Kinder und jugendliche Patienten länger als 1 Jahr und 2529 Kinder und jugendliche Patienten länger als 6 Monate behandelt.
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Klinische Studien mit Kindern und Jugendlichen
Gründe für den Abbruch der Behandlung aufgrund von Nebenwirkungen in klinischen Studien mit Kindern und Jugendlichen
In akuten placebokontrollierten Studien bei Kindern und Jugendlichen brachen 3,0 % (48/1613) der Atomoxetin-Patienten und 1,4 % (13/945) der Placebo-Patienten die Behandlung wegen Nebenwirkungen ab. Bei allen Studien (einschließlich offener und Langzeitstudien) brachen 6,3 % der Patienten mit schneller Metabolisierung (EM) und 11,2 % der Patienten mit langsamer Metabolisierung (PM) die Behandlung wegen einer Nebenwirkung ab. Bei den mit STRATTERA behandelten Patienten Reizbarkeit (0,3 %, N = 5); Schläfrigkeit (0,3 %, N = 5); Aggression (0,2 %, N=4); Übelkeit (0,2 %, N = 4); Erbrechen (0,2 %, N=4); Bauchschmerzen (0,2 %, N = 4); Verstopfung (0,1 %, N=2); Müdigkeit (0,1 %, N=2); Unwohlsein (0,1 %, N=2); und Kopfschmerzen (0,1 %, N = 2) waren die Gründe für den Abbruch, die von mehr als 1 Patient angegeben wurden.
Krampfanfälle
STRATTERA wurde bei pädiatrischen Patienten mit Anfallsleiden nicht systematisch untersucht, da diese Patienten während der Premarket-Tests des Produkts von klinischen Studien ausgeschlossen wurden. Im klinischen Entwicklungsprogramm wurden Krampfanfälle bei 0,2 % (12/5073) der Kinder mit einem Durchschnittsalter von 10 Jahren (Bereich 6 bis 16 Jahre) berichtet. In diesen klinischen Studien betrug das Anfallsrisiko bei langsamen Metabolisierern 0,3 % (1/293) im Vergleich zu 0,2 % (11/4741) bei schnellen Metabolisierern.
Häufig beobachtete Nebenwirkungen in Placebo-kontrollierten Studien bei akuten Kindern und Jugendlichen
Häufig beobachtete Nebenwirkungen im Zusammenhang mit der Anwendung von STRATTERA (Inzidenz von 2 % oder mehr) und nicht mit gleicher Inzidenz bei mit Placebo behandelten Patienten (Inzidenz von STRATTERA 40 mg höher als Placebo) sind in Tabelle 2 aufgeführt. Die Ergebnisse waren bei der BID ähnlich und die QD-Studie, außer wie in Tabelle 3 gezeigt, die sowohl BID- als auch QD-Ergebnisse für ausgewählte Nebenwirkungen basierend auf statistisch signifikanten Breslow-Day-Tests zeigt. Die am häufigsten beobachteten Nebenwirkungen bei Patienten, die mit STRATTERA behandelt wurden (Inzidenz von 5 % oder mehr und mindestens doppelt so häufig wie bei Placebo-Patienten, entweder bei BID- oder QD-Dosierung) waren: Übelkeit, Erbrechen, Müdigkeit, verminderter Appetit, Bauchschmerzen und Somnolenz (siehe Tabellen 2 und 3).
Zusätzliche Daten aus klinischen ADHS-Studien (kontrolliert und unkontrolliert) haben gezeigt, dass bei etwa 5 bis 10 % der pädiatrischen Patienten potenziell klinisch bedeutsame Veränderungen der Herzfrequenz (≥ 20 Schläge pro Minute) oder des Blutdrucks (≥ 15 bis 20 mmHg) auftraten [siehe KONTRAINDIKATIONEN und WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Die folgenden Nebenwirkungen traten bei mindestens 2 % der kindlichen und jugendlichen CYP2D6-PM-Patienten auf und waren bei PM-Patienten statistisch signifikant häufiger als bei CYP2D6-EM-Patienten: Schlaflosigkeit (11 % der PMs, 6 % der EMs); Gewicht verringert (7 % der PMs, 4 % der EMs); Verstopfung (7 % der PMs, 4 % der EMs); Depression1 (7 % der PMs, 4 % der EMs); Zittern (5 % der PMs, 1 % der EMs); Excoriation (4 % der PMs, 2 % der EMs); mittlere Schlaflosigkeit (3 % der PMs, 1 % der EMs); Konjunktivitis (3 % der PMs, 1 % der EMs); Synkope (3 % der PMs, 1 % der EMs); frühes Erwachen am Morgen (2 % der PMs, 1 % der EMs); Mydriasis (2 % der PMs, 1 % der EMs); Sedierung (4 % der PMs, 2 % der EMs).
1Depression umfasst die folgenden Begriffe: Depression, Major Depression, depressive Symptome, depressive Stimmung, Dysphorie.
Klinische Studien für Erwachsene
Gründe für den Abbruch der Behandlung aufgrund von Nebenwirkungen in placebokontrollierten Studien mit akuten Erwachsenen
In den akuten Placebo-kontrollierten Studien mit Erwachsenen brachen 11,3 % (61/541) der Atomoxetin-Patienten und 3,0 % (12/405) der Placebo-Patienten die Behandlung wegen Nebenwirkungen ab. Bei den mit STRATTERA behandelten Patienten traten Schlaflosigkeit (0,9 %, N=5); Übelkeit (0,9 %, N = 5); Brustschmerzen (0,6 %, N = 3); Müdigkeit (0,6 %, N=3); Angst (0,4 %, N=2); erektile Dysfunktion (0,4 %, N=2); Stimmungsschwankungen (0,4 %, N=2); Nervosität (0,4 %, N=2); Herzklopfen (0,4 %, N=2); und Harnverhalt (0,4 %, N = 2) waren die Gründe für den Abbruch, die von mehr als 1 Patient angegeben wurden.
Krampfanfälle
STRATTERA wurde bei erwachsenen Patienten mit Anfallsleiden nicht systematisch untersucht, da diese Patienten während der Premarket-Tests des Produkts von klinischen Studien ausgeschlossen wurden. Im klinischen Entwicklungsprogramm wurden bei 0,1 % (1/748) der erwachsenen Patienten Krampfanfälle gemeldet. In diesen klinischen Studien berichtete kein langsamer Metabolisierer (0/43) über Anfälle, verglichen mit 0,1 % (1/705) bei schnellen Metabolisierern.
Häufig beobachtete Nebenwirkungen in akuten Placebo-kontrollierten Studien mit Erwachsenen
Häufig beobachtete Nebenwirkungen im Zusammenhang mit der Anwendung von STRATTERA (Inzidenz von 2 % oder mehr) und nicht mit gleicher Inzidenz bei mit Placebo behandelten Patienten (Inzidenz von STRATTERA 25 mg höher als bei Placebo) sind in Tabelle 4 aufgeführt. Die am häufigsten beobachteten Nebenwirkungen bei Patienten, die mit STRATTERA behandelt wurden (Inzidenz von 5 % oder mehr und mindestens doppelt so häufig wie bei Placebo-Patienten): Verstopfung, Mundtrockenheit, Übelkeit, verminderter Appetit, Schwindel, erektile Dysfunktion und verzögertes Wasserlassen (siehe Tabelle 4). Zusätzliche Daten aus klinischen ADHS-Studien (kontrolliert und unkontrolliert) haben gezeigt, dass bei ungefähr 5 bis 10 % der erwachsenen Patienten potenziell klinisch bedeutsame Veränderungen der Herzfrequenz (≥ 20 Schläge pro Minute) oder des Blutdrucks (≥ 15 bis 20 mmHg) auftraten [siehe KONTRAINDIKATIONEN und WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Die folgenden unerwünschten Ereignisse traten bei mindestens 2 % der erwachsenen CYP2D6-Patienten mit langsamem Metabolisierer (PM) auf und waren statistisch signifikant häufiger bei PM-Patienten im Vergleich zu CYP2D6-Patienten mit schnellem Metabolisierer (EM): verschwommenes Sehen (4 % der PMs, 1 % der EMs). ); trockener Mund (35 % der PMs, 17 % der EMs); Verstopfung (11 % der PMs, 7 % der EMs); Nervosität (5 % der PMs, 2 % der EMs); verminderter Appetit (23 % der PMs, 15 % der EMs); Zittern (5 % der PMs, 1 % der EMs); Schlaflosigkeit (19 % der PMs, 11 % der EMs); Schlafstörung (7 % der PMs, 3 % der EMs); mittlere Schlaflosigkeit (5 % der PMs, 3 % der EMs); unheilbare Schlaflosigkeit (3 % der PMs, 1 % der EMs); Harnverhalt (6 % der PMs, 1 % der EMs); erektile Dysfunktion (21 % der PMs, 9 % der EMs); Ejakulationsstörung (6 % der PMs, 2 % der EMs); Hyperhidrose (15 % der PMs, 7 % der EMs); periphere Kälte (3 % der PMs, 1 % der EMs).
Männliche und weibliche sexuelle Dysfunktion
Atomoxetin scheint bei einigen Patienten die sexuelle Funktion zu beeinträchtigen. Veränderungen des sexuellen Verlangens, der sexuellen Leistung und der sexuellen Befriedigung werden in den meisten klinischen Studien nicht gut bewertet, weil sie besonderer Aufmerksamkeit bedürfen und weil Patienten und Ärzte möglicherweise zögern, darüber zu sprechen. Dementsprechend ist es wahrscheinlich, dass Schätzungen der Häufigkeit von unerwünschten sexuellen Erfahrungen und Verhaltensweisen, die in der Produktkennzeichnung angegeben werden, die tatsächliche Häufigkeit unterschätzen. Tabelle 4 oben zeigt die Inzidenz sexueller Nebenwirkungen, die von mindestens 2 % der erwachsenen Patienten, die STRATTERA in placebokontrollierten Studien einnahmen, berichtet wurden.
Es gibt keine adäquaten und gut kontrollierten Studien, die sexuelle Funktionsstörungen bei der Behandlung mit STRATTERA 40 mg untersuchten. Obwohl es schwierig ist, das genaue Risiko einer sexuellen Funktionsstörung im Zusammenhang mit der Anwendung von STRATTERA 40 mg abzuschätzen, sollten sich Ärzte routinemäßig nach solchen möglichen Nebenwirkungen erkundigen.
Postmarketing-Spontanberichte
Die folgenden Nebenwirkungen wurden während der Anwendung von STRATTERA nach der Zulassung festgestellt. Sofern nicht anders angegeben, sind diese Nebenwirkungen bei Erwachsenen und Kindern und Jugendlichen aufgetreten. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Herz-Kreislauf-System - QT-Verlängerung, Synkope.
Periphere vaskuläre Effekte - Raynaud-Phänomen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort - Lethargie.
Bewegungsapparat - Rhabdomyolyse.
Erkrankungen des Nervensystems - Hypästhesie; Parästhesien bei Kindern und Jugendlichen; sensorische Störungen; Tics.
Psychische Störungen - Depression und depressive Stimmung; Angst, Libido verändert sich.
Krampfanfälle In der Zeit nach Markteinführung wurden Krampfanfälle gemeldet. Zu den Anfallsfällen nach der Markteinführung gehören Patienten mit vorbestehenden Anfallsleiden und solche mit identifizierten Risikofaktoren für Anfälle sowie Patienten, bei denen weder in der Vorgeschichte noch Risikofaktoren für Anfälle identifiziert wurden. Die genaue Beziehung zwischen STRATTERA 40 mg und Krampfanfällen ist aufgrund der Ungewissheit über das Hintergrundrisiko von Krampfanfällen bei ADHS-Patienten schwer zu beurteilen.
Erkrankungen der Haut und des Unterhautgewebes - Alopezie, Hyperhidrose.
Urogenitalsystem - Beckenschmerzen bei Männern; Harnverzögerung bei Kindern und Jugendlichen; Harnverhalt bei Kindern und Jugendlichen.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Monoaminoxidase-Inhibitoren
Bei anderen Arzneimitteln, die die Monoaminkonzentrationen im Gehirn beeinflussen, liegen Berichte über schwerwiegende, manchmal tödliche Reaktionen vor (einschließlich Hyperthermie, Starrheit, Myoklonus, autonome Instabilität mit möglichen schnellen Schwankungen der Vitalfunktionen und Veränderungen des mentalen Status, einschließlich extremer Erregung bis hin zu Delirium und Koma). ) bei Einnahme in Kombination mit einem MAOI. Einige Fälle zeigten Merkmale, die dem malignen neuroleptischen Syndrom ähneln. Solche Reaktionen können auftreten, wenn diese Arzneimittel gleichzeitig oder in unmittelbarer Nähe gegeben werden [siehe KONTRAINDIKATIONEN ].
Wirkung von CYP2D6-Inhibitoren auf Atomoxetin
Bei schnellen Metabolisierern (EMs) erhöhen Inhibitoren von CYP2D6 (z. B. Paroxetin, Fluoxetin und Chinidin) die Steady-State-Plasmakonzentrationen von Atomoxetin auf ähnliche Expositionen wie bei langsamen Metabolisierern (PMs). Bei EM-Personen, die mit Paroxetin oder Fluoxetin behandelt werden, ist die AUC von Atomoxetin etwa 6- bis 8-fach und Css,max etwa 3- bis 4-fach größer als die von Atomoxetin allein.
In-vitro-Studien deuten darauf hin, dass die gleichzeitige Verabreichung von Cytochrom-P450-Inhibitoren an PMs die Plasmakonzentrationen von Atomoxetin nicht erhöht.
Antihypertensiva und blutdrucksenkende Mittel
Wegen möglicher Auswirkungen auf den Blutdruck sollte STRATTERA mit Vorsicht zusammen mit blutdrucksenkenden Arzneimitteln und blutdruckerhöhenden Mitteln (z. B. Dopamin, Dobutamin) oder anderen blutdrucksteigernden Arzneimitteln angewendet werden.
Alberol
STRATTERA sollte Patienten, die mit systemisch verabreichtem (oral oder intravenös) Albuterol (oder anderen Beta2-Agonisten) behandelt werden, mit Vorsicht verabreicht werden, da die Wirkung von Albuterol auf das Herz-Kreislauf-System verstärkt werden kann, was zu einem Anstieg der Herzfrequenz und des Blutdrucks führen kann. Albuterol (600 mcg iv über 2 Stunden) führte zu einem Anstieg der Herzfrequenz und des Blutdrucks. Diese Wirkungen wurden durch Atomoxetin (60 mg BID für 5 Tage) verstärkt und waren am ausgeprägtesten nach der anfänglichen gleichzeitigen Verabreichung von Albuterol und Atomoxetin. Diese Wirkungen auf die Herzfrequenz und den Blutdruck wurden jedoch in einer anderen Studie nach gleichzeitiger Verabreichung einer inhalativen Dosis von Albuterol (200-800 mcg) und Atomoxetin (80 mg einmal täglich für 5 Tage) bei 21 gesunden asiatischen Probanden, die wegen schlechter Behandlung ausgeschlossen wurden, nicht beobachtet Metabolisiererstatus.
Wirkung von Atomoxetin auf P450-Enzyme
Atomoxetin verursachte keine klinisch bedeutsame Hemmung oder Induktion von Cytochrom-P450-Enzymen, einschließlich CYP1A2, CYP3A, CYP2D6 und CYP2C9.
CYP3A-Substrat (z. B. Midazolam)
Die gleichzeitige Verabreichung von STRATTERA (60 mg zweimal täglich für 12 Tage) mit Midazolam, einer Modellverbindung für Arzneimittel, die durch CYP3A4 metabolisiert werden (Einzeldosis von 5 mg), führte zu einem Anstieg der AUC von Midazolam um 15 %. Für Arzneimittel, die durch CYP3A metabolisiert werden, wird keine Dosisanpassung empfohlen.
CYP2D6-Substrat (z. B. Desipramin)
Die gleichzeitige Verabreichung von STRATTERA (40 oder 60 mg zweimal täglich über 13 Tage) mit Desipramin, einer Modellverbindung für CYP2D6-metabolisierte Arzneimittel (Einzeldosis von 50 mg), veränderte die Pharmakokinetik von Desipramin nicht. Für Arzneimittel, die durch CYP2D6 metabolisiert werden, wird keine Dosisanpassung empfohlen.
Alkohol
Der Konsum von Ethanol mit STRATTERA 18 mg veränderte die berauschende Wirkung von Ethanol nicht.
Methylphenidat
Die gleichzeitige Verabreichung von Methylphenidat mit STRATTERA 18 mg verstärkte die kardiovaskulären Wirkungen nicht über die mit Methylphenidat allein beobachteten Wirkungen hinaus.
Arzneimittel, die stark an Plasmaproteine gebunden sind
In-vitro-Studien zur Arzneimittelverdrängung wurden mit Atomoxetin und anderen stark gebundenen Arzneimitteln in therapeutischen Konzentrationen durchgeführt. Atomoxetin hatte keinen Einfluss auf die Bindung von Warfarin, Acetylsalicylsäure, Phenytoin oder Diazepam an Humanalbumin. In ähnlicher Weise beeinflussten diese Verbindungen die Bindung von Atomoxetin an Humanalbumin nicht.
Medikamente, die den pH-Wert des Magens beeinflussen
Arzneimittel, die den Magen-pH-Wert erhöhen (Magnesiumhydroxid/Aluminiumhydroxid, Omeprazol) hatten keinen Einfluss auf die Bioverfügbarkeit von STRATTERA.
Drogenmissbrauch und -abhängigkeit
Kontrollierte Substanz
STRATTERA ist keine kontrollierte Substanz.
Missbrauch
In einer randomisierten, doppelblinden, placebokontrollierten Studie zum Missbrauchspotenzial bei Erwachsenen, in der die Wirkungen von STRATTERA und Placebo verglichen wurden, war STRATTERA nicht mit einem Reaktionsmuster verbunden, das auf stimulierende oder euphorisierende Eigenschaften hindeutete.
Abhängigkeit
Klinische Studiendaten bei über 2000 Kindern, Jugendlichen und Erwachsenen mit ADHS und über 1200 Erwachsenen mit Depressionen zeigten nur vereinzelte Vorfälle von Medikamentenabzweigung oder unsachgemäßer Selbstverabreichung im Zusammenhang mit STRATTERA. Es gab keine Hinweise auf ein Wiederauftreten der Symptome oder Nebenwirkungen, die auf ein Absetzen des Arzneimittels oder ein Entzugssyndrom hindeuteten.
Tierische Erfahrung
Studien zur Arzneimitteldiskriminierung bei Ratten und Affen zeigten eine inkonsistente Stimulus-Generalisierung zwischen Atomoxetin und Kokain.
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Suizidgedanken
STRATTERA erhöhte das Risiko von Suizidgedanken in Kurzzeitstudien bei Kindern und Jugendlichen mit Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS). Gepoolte Analysen von Placebo-kontrollierten Kurzzeitstudien (6 bis 18 Wochen) mit STRATTERA bei Kindern und Jugendlichen haben ein höheres Risiko für Suizidgedanken zu einem frühen Zeitpunkt der Behandlung bei denjenigen gezeigt, die STRATTERA erhielten. Es gab insgesamt 12 Studien (11 zu ADHS und 1 zu Enuresis) mit mehr als 2200 Patienten (einschließlich 1357 Patienten, die STRATTERA erhielten, und 851, die Placebo erhielten). Das durchschnittliche Risiko für Suizidgedanken bei Patienten, die STRATTERA erhielten, betrug 0,4 % (5/1357 Patienten), verglichen mit keinem Risiko bei den mit Placebo behandelten Patienten. Unter diesen etwa 2200 Patienten gab es 1 Suizidversuch bei einem mit STRATTERA behandelten Patienten. In diesen Studien kam es zu keinen Suiziden. Alle Reaktionen traten bei Kindern im Alter von 12 Jahren oder jünger auf. Alle Reaktionen traten während des ersten Behandlungsmonats auf. Es ist nicht bekannt, ob sich das Risiko von Suizidgedanken bei pädiatrischen Patienten auch auf eine längerfristige Anwendung erstreckt. Eine ähnliche Analyse bei erwachsenen Patienten, die entweder wegen ADHS oder einer Major Depression (MDD) mit STRATTERA behandelt wurden, ergab kein erhöhtes Risiko für Suizidgedanken oder -verhalten im Zusammenhang mit der Anwendung von STRATTERA.
Alle pädiatrischen Patienten, die mit STRATTERA behandelt werden, sollten angemessen überwacht und engmaschig auf eine klinische Verschlechterung, Suizidalität und ungewöhnliche Verhaltensänderungen beobachtet werden, insbesondere während der ersten Monate einer medikamentösen Therapie oder bei Dosisänderungen, entweder Erhöhungen oder Verringerungen .
Die folgenden Symptome wurden bei STRATTERA berichtet: Angst, Unruhe, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Unruhe), Hypomanie und Manie. Obwohl ein kausaler Zusammenhang zwischen dem Auftreten solcher Symptome und dem Auftreten suizidaler Impulse nicht hergestellt werden konnte, besteht die Sorge, dass solche Symptome Vorboten einer aufkommenden Suizidalität darstellen könnten. Daher sollten Patienten, die mit STRATTERA 40 mg behandelt werden, auf das Auftreten solcher Symptome hin beobachtet werden.
Bei Patienten, bei denen eine aufkommende Suizidalität oder Symptome auftreten, die Vorboten einer aufkommenden Suizidalität sein könnten, sollte eine Änderung des therapeutischen Schemas in Betracht gezogen werden, einschließlich eines möglichen Absetzens der Medikation, insbesondere wenn diese Symptome schwerwiegend oder plötzlich auftreten oder nicht Teil der Suizidalität waren die Symptome des Patienten.
Familien und Betreuer von pädiatrischen Patienten, die mit STRATTERA 10 mg behandelt werden, sollten auf die Notwendigkeit aufmerksam gemacht werden, Patienten auf das Auftreten von Unruhe, Reizbarkeit, ungewöhnlichen Verhaltensänderungen und den anderen oben beschriebenen Symptomen sowie auf das Auftreten von Suizidalität zu überwachen Melden Sie solche Symptome unverzüglich dem Gesundheitsdienstleister. Eine solche Überwachung sollte die tägliche Beobachtung durch Familien und Betreuer umfassen.
Schwere Leberschädigung
Postmarketing-Berichte weisen darauf hin, dass STRATTERA schwere Leberschäden verursachen kann. Obwohl in klinischen Studien mit etwa 6000 Patienten keine Hinweise auf eine Leberschädigung festgestellt wurden, gab es seltene Fälle von klinisch signifikanten Leberschädigungen, die nach der Markteinführung als wahrscheinlich oder möglicherweise mit der Anwendung von STRATTERA 40 mg zusammenhängend erachtet wurden. Seltene Fälle von Leberversagen wurden ebenfalls berichtet, darunter ein Fall, der zu einer Lebertransplantation führte. Aufgrund einer wahrscheinlichen Unterberichterstattung ist es unmöglich, eine genaue Schätzung der tatsächlichen Inzidenz dieser Reaktionen zu liefern. Gemeldete Fälle von Leberschäden traten in den meisten Fällen innerhalb von 120 Tagen nach Beginn der Atomoxetin-Behandlung auf, und einige Patienten zeigten deutlich erhöhte Leberenzyme [>20 x Obergrenze des Normalwerts (ULN)] und Gelbsucht mit signifikant erhöhten Bilirubinspiegeln (>2 X ULN), gefolgt von Erholung nach Absetzen von Atomoxetin. Bei einem Patienten trat die Leberschädigung, die sich durch erhöhte Leberenzyme bis zum 40-fachen des oberen Normwertes und Gelbsucht mit Bilirubin bis zum 12-fachen des oberen Normwertes manifestierte, bei einer erneuten Provokation erneut auf und erholte sich nach Absetzen des Medikaments, was den Nachweis erbrachte, dass STRATTERA wahrscheinlich die Leberschädigung verursachte. Solche Reaktionen können mehrere Monate nach Beginn der Therapie auftreten, aber Laboranomalien können sich noch mehrere Wochen nach Absetzen des Arzneimittels verschlechtern. Der oben beschriebene Patient erholte sich von seiner Leberverletzung und benötigte keine Lebertransplantation.
STRATTERA sollte bei Patienten mit Gelbsucht oder Laborbefunden einer Leberschädigung abgesetzt und nicht wieder aufgenommen werden. Labortests zur Bestimmung der Leberenzymwerte sollten beim ersten Symptom oder Anzeichen einer Leberfunktionsstörung (z. B. Juckreiz, dunkler Urin, Gelbsucht, Druckschmerz im rechten oberen Quadranten oder unerklärliche „grippeähnliche“ Symptome) durchgeführt werden [siehe Labortests , INFORMATIONEN ZUM PATIENTEN ].
Schwerwiegende kardiovaskuläre Ereignisse
Plötzlicher Tod und bereits bestehende strukturelle Herzfehler oder andere schwerwiegende Herzprobleme
Kinder und Jugendliche
Plötzlicher Herztod wurde im Zusammenhang mit der Behandlung mit Atomoxetin in üblichen Dosen bei Kindern und Jugendlichen mit strukturellen Herzanomalien oder anderen schweren Herzproblemen berichtet. Obwohl einige schwerwiegende Herzprobleme allein ein erhöhtes Risiko für einen plötzlichen Tod mit sich bringen, sollte Atomoxetin im Allgemeinen nicht bei Kindern oder Jugendlichen mit bekannten schwerwiegenden strukturellen Herzanomalien, Kardiomyopathie, schwerwiegenden Herzrhythmusstörungen oder anderen schwerwiegenden Herzproblemen angewendet werden, die sie einer erhöhten Anfälligkeit aussetzen können zu den noradrenergen Wirkungen von Atomoxetin.
Erwachsene
Plötzliche Todesfälle, Schlaganfall und Myokardinfarkt wurden bei Erwachsenen berichtet, die Atomoxetin in üblichen Dosen gegen ADHS einnahmen. Obwohl die Rolle von Atomoxetin in diesen Fällen bei Erwachsenen ebenfalls unbekannt ist, haben Erwachsene eine größere Wahrscheinlichkeit als Kinder, an schweren strukturellen Herzanomalien, Kardiomyopathie, schweren Herzrhythmusstörungen, koronarer Herzkrankheit oder anderen schweren Herzproblemen zu leiden. Es sollte erwogen werden, Erwachsene mit klinisch signifikanten Herzfehlern nicht zu behandeln.
Beurteilung des kardiovaskulären Status bei Patienten, die mit Atomoxetin behandelt werden
Kinder, Jugendliche oder Erwachsene, die für eine Behandlung mit Atomoxetin in Betracht gezogen werden, sollten eine sorgfältige Anamnese (einschließlich Beurteilung einer Familienanamnese mit plötzlichem Tod oder ventrikulärer Arrhythmie) und eine körperliche Untersuchung haben, um das Vorliegen einer Herzerkrankung zu beurteilen, und sollten weitere kardiologische Untersuchungen erhalten Auswertung, wenn die Befunde auf eine solche Erkrankung hindeuten (z. B. Elektrokardiogramm und Echokardiogramm). Patienten, die während der Behandlung mit Atomoxetin Symptome wie Brustschmerzen bei Belastung, unerklärliche Synkopen oder andere Symptome entwickeln, die auf eine Herzerkrankung hindeuten, sollten sich umgehend einer kardiologischen Untersuchung unterziehen.
Auswirkungen auf Blutdruck und Herzfrequenz
STRATTERA sollte mit Vorsicht bei Patienten angewendet werden, deren Grunderkrankungen sich durch einen Anstieg des Blutdrucks oder der Herzfrequenz verschlechtern könnten, wie z. B. bestimmte Patienten mit Bluthochdruck, Tachykardie oder kardiovaskulären oder zerebrovaskulären Erkrankungen. Es sollte nicht bei Patienten mit schweren Herz- oder Gefäßerkrankungen angewendet werden, von denen zu erwarten ist, dass sich ihr Zustand verschlechtert, wenn sie einen klinisch bedeutsamen Anstieg des Blutdrucks oder der Herzfrequenz erfahren [siehe KONTRAINDIKATIONEN ]. Puls und Blutdruck sollten zu Studienbeginn, nach Erhöhung der STRATTERA-Dosis und in regelmäßigen Abständen während der Therapie gemessen werden, um mögliche klinisch bedeutsame Anstiege zu erkennen.
Die folgende Tabelle enthält Daten aus placebokontrollierten Kurzzeitstudien aus klinischen Studien für die Anteile von Patienten mit einem Anstieg von: diastolischem Blutdruck ≥ 15 mm Hg; systolischer Blutdruck ≥20 mm Hg; Herzfrequenz größer oder gleich 20 bpm, sowohl bei Kindern als auch bei Erwachsenen (siehe Tabelle 1).
In placebokontrollierten Zulassungsstudien mit pädiatrischen Patienten wurde Tachykardie als unerwünschtes Ereignis bei 0,3 % (5/1597) dieser mit STRATTERA 10 mg behandelten Patienten identifiziert, verglichen mit 0 % (0/934) der Placebo-Patienten. Der mittlere Anstieg der Herzfrequenz bei Patienten mit schneller Metabolisierung (EM) betrug 5,0 Schläge/Minute und bei Patienten mit langsamer Metabolisierung (PM) 9,4 Schläge/Minute.
In klinischen Studien mit Erwachsenen, in denen der EM/PM-Status verfügbar war, war der mittlere Anstieg der Herzfrequenz bei PM-Patienten signifikant höher als bei EM-Patienten (11 Schläge/Minute gegenüber 7,5 Schlägen/Minute). Die Auswirkungen auf die Herzfrequenz könnten bei einigen PM-Patienten klinisch bedeutsam sein.
In placebokontrollierten Zulassungsstudien mit erwachsenen Patienten wurde bei 1,5 % (8/540) der mit STRATTERA 25 mg behandelten Patienten Tachykardie als Nebenwirkung festgestellt, verglichen mit 0,5 % (2/402) der Placebo-Patienten.
In klinischen Studien mit Erwachsenen, in denen der EM/PM-Status verfügbar war, war die mittlere Veränderung des diastolischen Blutdrucks gegenüber dem Ausgangswert bei PM-Patienten höher als bei EM-Patienten (4,21 gegenüber 2,13 mm Hg), ebenso wie die mittlere Veränderung des systolischen Blutdrucks (PM : 2,75 versus EM: 2,40 mm Hg). Die Blutdruckeffekte könnten bei einigen PM-Patienten klinisch bedeutsam sein.
Bei Patienten, die STRATTERA einnahmen, wurde über orthostatische Hypotonie und Synkope berichtet. In Zulassungsstudien für Kinder und Jugendliche traten bei 0,2 % (12/5596) der mit STRATTERA behandelten Patienten eine orthostatische Hypotonie und bei 0,8 % (46/5596) eine Synkope auf. In Kurzzeit-Zulassungsstudien bei Kindern und Jugendlichen trat bei 1,8 % (6/340) der mit STRATTERA behandelten Patienten eine orthostatische Hypotonie auf, verglichen mit 0,5 % (1/207) der mit Placebo behandelten Patienten. Synkopen wurden während der placebokontrollierten ADHS-Zulassungsstudien bei Kindern und Jugendlichen über einen kurzen Zeitraum nicht berichtet. STRATTERA 10 mg sollte bei allen Erkrankungen, die Patienten für Hypotonie prädisponieren könnten, oder bei Erkrankungen, die mit plötzlichen Herzfrequenz- oder Blutdruckänderungen einhergehen, mit Vorsicht angewendet werden.
Auftreten neuer psychotischer oder manischer Symptome
Behandlung Auftretende psychotische oder manische Symptome, z. B. Halluzinationen, wahnhaftes Denken oder Manie bei Kindern und Jugendlichen ohne Vorgeschichte einer psychotischen Erkrankung oder Manie, können durch Atomoxetin in üblichen Dosen verursacht werden. Wenn solche Symptome auftreten, sollte eine mögliche kausale Rolle von Atomoxetin in Betracht gezogen und ein Absetzen der Behandlung in Betracht gezogen werden. In einer gepoolten Analyse mehrerer placebokontrollierter Kurzzeitstudien traten solche Symptome bei etwa 0,2 % (4 Patienten mit Reaktionen von 1939, die mehrere Wochen lang Atomoxetin in üblichen Dosen erhielten) der mit Atomoxetin behandelten Patienten auf, verglichen mit 0 von 0 1056 mit Placebo behandelte Patienten.
Screening von Patienten auf bipolare Störungen
Im Allgemeinen sollte bei der Behandlung von ADHS bei Patienten mit komorbider bipolarer Störung besondere Vorsicht walten, da Bedenken hinsichtlich einer möglichen Induktion einer gemischten/manischen Episode bei Patienten mit einem Risiko für eine bipolare Störung bestehen. Ob eines der oben beschriebenen Symptome eine solche Konversion darstellt, ist nicht bekannt. Vor Beginn der Behandlung mit STRATTERA 40 mg sollten Patienten mit komorbiden depressiven Symptomen jedoch angemessen untersucht werden, um festzustellen, ob bei ihnen ein Risiko für eine bipolare Störung besteht; Ein solches Screening sollte eine detaillierte psychiatrische Vorgeschichte umfassen, einschließlich einer Familienanamnese von Selbstmord, bipolarer Störung und Depression.
Aggressives Verhalten oder Feindseligkeit
Patienten, die mit der Behandlung von ADHS beginnen, sollten auf das Auftreten oder die Verschlechterung von aggressivem Verhalten oder Feindseligkeit überwacht werden. Aggressives Verhalten oder Feindseligkeit wird häufig bei Kindern und Jugendlichen mit ADHS beobachtet. In pädiatrischen kontrollierten Kurzzeitstudien berichteten 21/1308 (1,6 %) der mit Atomoxetin behandelten Patienten im Vergleich zu 9/806 (1,1 %) der mit Placebo behandelten Patienten spontan über behandlungsbedingte feindselige unerwünschte Ereignisse (Gesamtrisikoverhältnis von 1,33 [95 % KI 0,67-2,64 – nicht statistisch signifikant]). In placebokontrollierten klinischen Studien mit Erwachsenen berichteten 6/1697 (0,35 %) der mit Atomoxetin behandelten Patienten gegenüber 4/1560 (0,26 %) der mit Placebo behandelten Patienten spontan über behandlungsbedingte feindselige unerwünschte Ereignisse (Gesamtrisikoverhältnis von 1,38 [95 % KI 0,39-4,88 – statistisch nicht signifikant]). Obwohl dies kein schlüssiger Beweis dafür ist, dass STRATTERA 10 mg aggressives Verhalten oder Feindseligkeit auslöst, wurden diese Verhaltensweisen in klinischen Studien bei Kindern, Jugendlichen und Erwachsenen, die mit STRATTERA behandelt wurden, häufiger beobachtet als mit Placebo.
Allergische Ereignisse
Obwohl gelegentlich, wurde bei Patienten, die STRATTERA einnahmen, über allergische Reaktionen berichtet, darunter anaphylaktische Reaktionen, angioneurotisches Ödem, Urtikaria und Hautausschlag.
Auswirkungen auf den Urinabfluss aus der Blase
In kontrollierten ADHS-Studien mit Erwachsenen waren die Raten von Harnverhalt (1,7 %, 9/540) und zögerlichem Wasserlassen (5,6 %, 30/540) bei Atomoxetin-Patienten im Vergleich zu Placebo-Patienten (0 %, 0/402; 0,5 %, 2/402). Zwei erwachsene Atomoxetin-Probanden und keine Placebo-Probanden brachen die kontrollierten klinischen Studien wegen Harnverhaltung ab. Eine Beschwerde über Harnverhalt oder Harnverhalt sollte als potenziell im Zusammenhang mit Atomoxetin angesehen werden.
Priapismus
Seltene Postmarketing-Fälle von Priapismus, definiert als schmerzhafte und nicht schmerzhafte Erektion des Penis, die länger als 4 Stunden anhält, wurden bei pädiatrischen und erwachsenen Patienten berichtet, die mit STRATTERA behandelt wurden. Die Erektionen verschwanden in Fällen, in denen Nachbeobachtungsinformationen verfügbar waren, einige nach Absetzen von STRATTERA. Bei Verdacht auf Priapismus ist eine sofortige ärztliche Behandlung erforderlich.
Auswirkungen auf das Wachstum
Die Daten zu den Langzeitwirkungen von STRATTERA 25 mg auf das Wachstum stammen aus offenen Studien, und Gewichts- und Größenänderungen werden mit normativen Bevölkerungsdaten verglichen. Im Allgemeinen bleibt die Gewichts- und Größenzunahme von mit STRATTERA behandelten pädiatrischen Patienten etwa in den ersten 9 bis 12 Behandlungsmonaten hinter den Prognosen aus normativen Bevölkerungsdaten zurück. Anschließend kommt es zu einer erneuten Gewichtszunahme, und nach etwa 3 Behandlungsjahren haben die mit STRATTERA behandelten Patienten durchschnittlich 17,9 kg zugenommen, 0,5 kg mehr als aufgrund ihrer Ausgangsdaten vorhergesagt. Nach etwa 12 Monaten stabilisiert sich die Zunahme der Körpergröße, und nach 3 Jahren haben die mit STRATTERA 40 mg behandelten Patienten durchschnittlich 19,4 cm zugenommen, 0,4 cm weniger als von ihren Ausgangsdaten vorhergesagt (siehe Abbildung 1 unten).
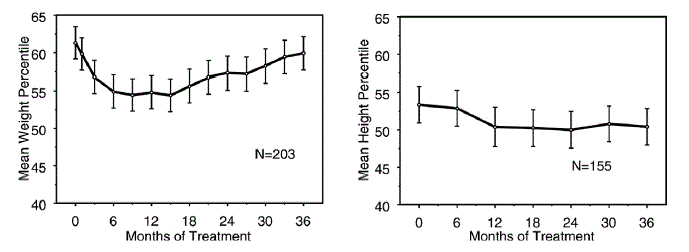
Abbildung 1: Durchschnittliche Gewichts- und Größenperzentile im Zeitverlauf für Patienten mit dreijähriger Behandlung mit STRATTERA 40 mg
Dieses Wachstumsmuster war im Allgemeinen unabhängig vom Pubertätsstatus zum Zeitpunkt des Behandlungsbeginns ähnlich. Patienten, die zu Beginn der Behandlung präpubertär waren (Mädchen ≤ 8 Jahre, Jungen ≤ 9 Jahre), nahmen nach drei Jahren durchschnittlich 2,1 kg und 1,2 cm weniger zu als vorhergesagt. Pubertätspatienten (Mädchen > 8 bis ≤ 13 Jahre, Jungen > 9 bis ≤ 14 Jahre) oder spätpubertär (Mädchen > 13 Jahre, Jungen > 14 Jahre) hatten eine durchschnittliche Gewichts- und Größenzunahme, die nahe bei oder lag übertraf die nach dreijähriger Behandlung vorhergesagten.
Das Wachstum folgte sowohl bei schnellen als auch bei langsamen Metabolisierern (EMs, PMs) einem ähnlichen Muster. PMs, die mindestens zwei Jahre lang behandelt wurden, nahmen durchschnittlich 2,4 kg und 1,1 cm weniger zu als vorhergesagt, während EMs durchschnittlich 0,2 kg und 0,4 cm weniger zunahmen als vorhergesagt.
In kontrollierten Kurzzeitstudien (bis zu 9 Wochen) verloren die mit STRATTERA behandelten Patienten durchschnittlich 0,4 kg und nahmen durchschnittlich 0,9 cm zu, verglichen mit einer Zunahme von 1,5 kg und 1,1 cm bei den mit Placebo behandelten Patienten. In einer kontrollierten Studie mit fester Dosis verloren 1,3 %, 7,1 %, 19,3 % und 29,1 % der Patienten in den Gruppen mit Placebo, 0,5, 1,2 und 1,8 mg/kg/Tag mindestens 3,5 % ihres Körpergewichts.
Das Wachstum sollte während der Behandlung mit STRATTERA überwacht werden.
Labortests
Routinelaboruntersuchungen sind nicht erforderlich.
CYP2D6-Metabolismus
Langsame Metabolisierer (PMs) von CYP2D6 haben eine 10-fach höhere AUC und eine 5-fach höhere Spitzenkonzentration bei einer gegebenen Dosis von STRATTERA 25 mg im Vergleich zu schnellen Metabolisierern (EMs). Ungefähr 7 % der kaukasischen Bevölkerung sind PMs. Labortests sind verfügbar, um CYP2D6-PMs zu identifizieren. Die Blutspiegel bei PMs ähneln denen, die durch die Einnahme starker Inhibitoren von CYP2D6 erreicht werden. Die höheren Blutspiegel in PMs führen zu einer höheren Rate einiger Nebenwirkungen von STRATTERA [siehe NEBENWIRKUNGEN ].
Gleichzeitige Anwendung von starken CYP2D6-Inhibitoren oder Anwendung bei Patienten, die bekanntermaßen CYP2D6-PMs sind
Atomoxetin wird hauptsächlich über den CYP2D6-Weg zu 4-Hydroxyatomoxetin metabolisiert. Bei gleichzeitiger Anwendung mit starken CYP2D6-Inhibitoren (z. B. Paroxetin, Fluoxetin und Chinidin) oder bei Verabreichung an CYP2D6-PMs kann eine Dosisanpassung von STRATTERA erforderlich sein. [Sehen DOSIERUNG UND ANWENDUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Informationen zur Patientenberatung
Siehe FDA-zugelassen INFORMATIONEN ZUM PATIENTEN .
Allgemeine Informationen
Ärzte sollten ihre Patienten anweisen, den Medikationsleitfaden vor Beginn der Therapie mit STRATTERA zu lesen und ihn jedes Mal erneut zu lesen, wenn die Verschreibung verlängert wird.
Verschreibende Ärzte oder andere Angehörige der Gesundheitsberufe sollten Patienten, ihre Familien und ihre Betreuer über die Vorteile und Risiken einer Behandlung mit STRATTERA informieren und sie bezüglich der angemessenen Anwendung beraten. Der verschreibende Arzt oder medizinisches Fachpersonal sollte Patienten, ihre Familien und ihre Betreuer anweisen, den Medikationsleitfaden zu lesen, und ihnen helfen, seinen Inhalt zu verstehen. Den Patienten sollte Gelegenheit gegeben werden, den Inhalt des Medikationsleitfadens zu diskutieren und Antworten auf eventuelle Fragen zu erhalten.
Die Patienten sollten auf die folgenden Probleme hingewiesen und gebeten werden, ihren verschreibenden Arzt zu informieren, wenn diese während der Einnahme von STRATTERA auftreten.
Selbstmordrisiko
Patienten, ihre Familien und ihre Betreuer sollten ermutigt werden, auf das Auftreten von Angstzuständen, Unruhe, Panikattacken, Schlaflosigkeit, Reizbarkeit, Feindseligkeit, Aggressivität, Impulsivität, Akathisie (psychomotorische Unruhe), Hypomanie, Manie und anderen ungewöhnlichen Verhaltensänderungen zu achten , Depression und Suizidgedanken, insbesondere zu Beginn der STRATTERA-Behandlung und bei Dosisanpassung. Familien und Betreuer von Patienten sollten darauf hingewiesen werden, täglich auf das Auftreten solcher Symptome zu achten, da Änderungen plötzlich auftreten können. Solche Symptome sollten dem verschreibenden Arzt oder medizinischen Fachpersonal des Patienten gemeldet werden, insbesondere wenn sie schwerwiegend sind, abrupt einsetzen oder nicht zu den Symptomen des Patienten gehörten. Symptome wie diese können mit einem erhöhten Risiko für Suizidgedanken und -verhalten verbunden sein und weisen auf die Notwendigkeit einer sehr engmaschigen Überwachung und möglicherweise einer Änderung der Medikation hin.
Schwere Leberschädigung
Patienten, die mit STRATTERA beginnen, sollten darauf hingewiesen werden, dass sich eine schwere Leberschädigung entwickeln kann. Die Patienten sollten angewiesen werden, sich sofort an ihren Arzt zu wenden, wenn sie Juckreiz, dunklen Urin, Gelbsucht, Druckschmerz im rechten oberen Quadranten oder unerklärliche „grippeähnliche“ Symptome entwickeln [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Aggression oder Feindseligkeit
Die Patienten sollten angewiesen werden, so schnell wie möglich ihren Arzt anzurufen, wenn sie eine Zunahme der Aggression oder Feindseligkeit bemerken.
Priapismus
Seltene Postmarketing-Fälle von Priapismus, definiert als schmerzhafte und nicht schmerzhafte Erektion des Penis, die länger als 4 Stunden anhält, wurden bei pädiatrischen und erwachsenen Patienten berichtet, die mit STRATTERA behandelt wurden. Die Eltern oder Erziehungsberechtigten von pädiatrischen Patienten, die STRATTERA 25 mg einnehmen, und von erwachsenen Patienten, die STRATTERA einnehmen, sollten darüber informiert werden, dass Priapismus eine sofortige ärztliche Behandlung erfordert.
Augenreizend
STRATTERA ist ein Augenreizmittel. STRATTERA-Kapseln sind nicht zum Öffnen bestimmt. Bei Kontakt des Kapselinhalts mit dem Auge ist das betroffene Auge sofort mit Wasser zu spülen und ärztlicher Rat einzuholen. Hände und alle möglicherweise kontaminierten Oberflächen sollten so schnell wie möglich gewaschen werden.
Arzneimittelwechselwirkung
Die Patienten sollten angewiesen werden, einen Arzt zu konsultieren, wenn sie verschreibungspflichtige oder rezeptfreie Arzneimittel, Nahrungsergänzungsmittel oder pflanzliche Heilmittel einnehmen oder einnehmen möchten.
Schwangerschaft
Patienten sollten angewiesen werden, einen Arzt zu konsultieren, wenn sie stillen, schwanger sind oder beabsichtigen, während der Einnahme von STRATTERA schwanger zu werden.
Essen
Die Patienten können STRATTERA 25 mg mit oder ohne Nahrung einnehmen.
Verpasste Dosis
Wenn Patienten eine Dosis vergessen haben, sollten sie angewiesen werden, diese so bald wie möglich einzunehmen, aber sie sollten innerhalb von 24 Stunden nicht mehr als die verschriebene Tagesgesamtdosis von STRATTERA einnehmen.
Beeinträchtigung der psychomotorischen Leistung
Die Patienten sollten angewiesen werden, beim Autofahren oder beim Bedienen gefährlicher Maschinen vorsichtig zu sein, bis sie hinreichend sicher sind, dass Atomoxetin ihre Leistungsfähigkeit nicht beeinträchtigt.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Atomoxetin-HCl war bei Ratten und Mäusen nicht karzinogen, wenn es 2 Jahre lang mit der Nahrung in zeitgewichteten Durchschnittsdosen von bis zu 47 bzw. 458 mg/kg/Tag verabreicht wurde. Die höchste Dosis, die bei Ratten verwendet wird, beträgt ungefähr das 8- bzw. 5-fache der maximalen Humandosis bei Kindern bzw. Erwachsenen, auf mg/m2-Basis. Die Plasmaspiegel (AUC) von Atomoxetin bei dieser Dosis bei Ratten werden auf das 1,8-fache (schnelle Metabolisierer) oder 0,2-fache (langsame Metabolisierer) derjenigen beim Menschen geschätzt, der die maximale Humandosis erhält. Die höchste Dosis, die bei Mäusen verwendet wird, beträgt etwa das 39- bzw. 26-fache der maximalen Humandosis bei Kindern bzw. Erwachsenen, auf mg/m2-Basis.
Mutagenese
Atomoxetin-HCl war in einer Reihe von Genotoxizitätsstudien negativ, darunter ein Reverse-Point-Mutation-Assay (Ames-Test), ein In-vitro-Maus-Lymphom-Assay, ein Chromosomenaberrationstest in Eierstockzellen des chinesischen Hamsters, ein außerplanmäßiger DNA-Synthesetest in Rattenhepatozyten und ein In-vivo-Mikrokerntest bei Mäusen. Es gab jedoch einen leichten Anstieg im Prozentsatz der Ovarialzellen des chinesischen Hamsters mit Diplochromosomen, was auf eine Endoreduplikation (numerische Aberration) hindeutet.
Der Metabolit N-Desmethylatomoxetin-HCl war im Ames-Test, Maus-Lymphom-Assay und außerplanmäßigen DNA-Synthesetest negativ.
Beeinträchtigung der Fruchtbarkeit
Atomoxetin-HCl beeinträchtigte die Fertilität von Ratten nicht, wenn es mit dem Futter in Dosen von bis zu 57 mg/kg/Tag verabreicht wurde, was etwa dem 6-fachen der maximalen Humandosis auf mg/m2-Basis entspricht.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschaftskategorie C
Trächtige Kaninchen wurden während der Organogenese mit bis zu 100 mg/kg/Tag Atomoxetin per Schlundsonde behandelt. Bei dieser Dosis wurde in 1 von 3 Studien eine Abnahme der lebenden Föten und eine Zunahme der frühen Resorptionen beobachtet. Es wurde ein leichter Anstieg der Inzidenzen von atypischem Ursprung der Halsschlagader und fehlender A. subclavia beobachtet. Diese Befunde wurden bei Dosen beobachtet, die eine leichte maternale Toxizität verursachten. Die Dosis ohne Wirkung für diese Befunde betrug 30 mg/kg/Tag. Die Dosis von 100 mg/kg entspricht etwa dem 23-fachen der maximalen Humandosis auf mg/m2-Basis; Plasmaspiegel (AUC) von Atomoxetin in dieser Dosis bei Kaninchen werden auf das 3,3-fache (schnelle Metabolisierer) oder 0,4-fache (langsame Metabolisierer) derjenigen beim Menschen geschätzt, der die maximale Humandosis erhält.
Ratten wurden 2 Wochen (Weibchen) oder 10 Wochen (Männchen) vor der Paarung mit bis zu etwa 50 mg/kg/Tag Atomoxetin (etwa das 6-fache der maximalen Humandosis auf mg/m2-Basis) im Futter behandelt Perioden der Organogenese und Laktation. In 1 von 2 Studien wurde eine Verringerung des Gewichts der Jungtiere und der Überlebensrate der Jungtiere beobachtet. Das verringerte Überleben der Jungtiere wurde auch bei 25 mg/kg (aber nicht bei 13 mg/kg) beobachtet. In einer Studie, in der Ratten ab 2 Wochen (Weibchen) bzw. 10 Wochen (Männchen) vor der Paarung mit Atomoxetin im Futter behandelt wurden, kam es während der gesamten Organogenese zu einer Abnahme des fötalen Gewichts (nur bei Weibchen) und einem Anstieg der Inzidenz von Eine unvollständige Ossifikation des Wirbelbogens bei Föten wurde bei 40 mg/kg/Tag (etwa das 5-fache der maximalen Humandosis auf mg/m2-Basis) beobachtet, jedoch nicht bei 20 mg/kg/Tag.
Es wurden keine unerwünschten Wirkungen auf den Fötus beobachtet, wenn trächtige Ratten während der Organogenese mit bis zu 150 mg/kg/Tag (etwa das 17-fache der maximalen Humandosis auf mg/m2-Basis) per Schlundsonde behandelt wurden. Es wurden keine angemessenen und gut kontrollierten Studien bei schwangeren Frauen durchgeführt. STRATTERA 18 mg sollte während der Schwangerschaft nicht angewendet werden, es sei denn, der potenzielle Nutzen rechtfertigt das potenzielle Risiko für den Fötus.
Arbeit und Lieferung
Die Geburt bei Ratten wurde durch Atomoxetin nicht beeinflusst. Die Wirkung von STRATTERA auf Wehen und Entbindung beim Menschen ist nicht bekannt.
Stillende Mutter
Atomoxetin und/oder seine Metaboliten wurden in die Milch von Ratten ausgeschieden. Es ist nicht bekannt, ob Atomoxetin in die Muttermilch übergeht. Vorsicht ist geboten, wenn STRATTERA einer stillenden Frau verabreicht wird.
Pädiatrische Verwendung
Jeder, der die Anwendung von STRATTERA bei einem Kind oder Jugendlichen in Betracht zieht, muss die potenziellen Risiken mit der klinischen Notwendigkeit abwägen [siehe KASTENWARNUNG und WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Pharmakokinetik von Atomoxetin bei Kindern und Jugendlichen ist ähnlich wie bei Erwachsenen. Die Sicherheit, Wirksamkeit und Pharmakokinetik von STRATTERA bei pädiatrischen Patienten unter 6 Jahren wurde nicht untersucht.
An jungen Ratten wurde eine Studie durchgeführt, um die Wirkungen von Atomoxetin auf das Wachstum und die neurologische und sexuelle Entwicklung zu bewerten. Ratten wurden ab der frühen postnatalen Phase (Tag 10 Jahre) bis ins Erwachsenenalter. Leichte Verzögerungen beim Einsetzen der vaginalen Durchgängigkeit (alle Dosen) und Präputialtrennung (10 und 50 mg/kg), leichte Abnahme des Nebenhodengewichts und der Spermienzahl (10 und 50 mg/kg) und eine leichte Abnahme der Corpora lutea (50 mg/kg). /kg) wurden beobachtet, aber es gab keine Auswirkungen auf die Fruchtbarkeit oder Fortpflanzungsfähigkeit. Bei 50 mg/kg wurde eine leichte Verzögerung des Ausbruchs der Schneidezähne beobachtet. Eine leichte Zunahme der motorischen Aktivität wurde am 15. Tag (Männer bei 10 und 50 mg/kg und Frauen bei 50 mg/kg) und am 30. Tag (Frauen bei 50 mg/kg) beobachtet, jedoch nicht am 60. Lebenstag. Es gab keine Auswirkungen auf Lern- und Gedächtnistests. Die Bedeutung dieser Befunde für den Menschen ist unbekannt.
Geriatrische Verwendung
Die Sicherheit, Wirksamkeit und Pharmakokinetik von STRATTERA 10 mg bei geriatrischen Patienten wurde nicht untersucht.
Leberinsuffizienz
Die Atomoxetin-Exposition (AUC) ist im Vergleich zu gesunden Probanden bei EM-Probanden mit mäßiger (Child-Pugh-Klasse B) (2-facher Anstieg) und schwerer (Child-Pugh-Klasse C) (4-facher Anstieg) Leberinsuffizienz erhöht. Bei Patienten mit mittelschwerer oder schwerer Leberinsuffizienz wird eine Dosisanpassung empfohlen [siehe DOSIERUNG UND ANWENDUNG ].
Niereninsuffizienz
EM-Patienten mit Nierenerkrankungen im Endstadium hatten eine höhere systemische Atomoxetin-Exposition als gesunde Probanden (ca. 65 % Anstieg), aber es gab keinen Unterschied, wenn die Exposition auf die mg/kg-Dosis korrigiert wurde. STRATTERA 25 mg kann daher ADHS-Patienten mit einer Nierenerkrankung im Endstadium oder geringerer Niereninsuffizienz unter Verwendung des normalen Dosierungsschemas verabreicht werden.
Geschlecht
Das Geschlecht hatte keinen Einfluss auf die Atomoxetin-Disposition.
Ethnischer Ursprung
Die ethnische Herkunft hatte keinen Einfluss auf die Atomoxetin-Disposition (mit der Ausnahme, dass PMs häufiger bei Kaukasiern auftreten).
Patienten mit Begleiterkrankungen
Tics bei Patienten mit ADHS und komorbidem Tourette-Syndrom
Atomoxetin, das in einem flexiblen Dosisbereich von 0,5 bis 1,5 mg/kg/Tag (mittlere Dosis von 1,3 mg/kg/Tag) verabreicht wurde, und Placebo wurden bei 148 randomisierten pädiatrischen Probanden (Alter 717 Jahre) mit einer DSM-IV-Diagnose von ADHS und verglichen komorbide Tic-Störung in einer 18-wöchigen, doppelblinden, placebokontrollierten Studie, in der die Mehrheit (80 %) an dieser Studie mit Tourette-Krankheit teilnahmen (Tourette-Krankheit: 116 Probanden; chronische motorische Tic-Störung: 29 Probanden). Eine Nicht-Unterlegenheitsanalyse ergab, dass STRATTERA 10 mg die Tics bei diesen Patienten nicht verschlimmerte, wie anhand des Yale Global Tic Severity Scale Total Score (YGTSS) bestimmt wurde. Von 148 Patienten, die in die akute Behandlungsphase eintraten, brachen 103 (69,6 %) Patienten die Studie ab. Der Hauptgrund für das Absetzen sowohl in der Atomoxetin- (38 von 76 Patienten, 50,0 %) als auch in der Placebo-Behandlungsgruppe (45 von 72 Patienten, 62,5 %) wurde als mangelnde Wirksamkeit identifiziert, wobei die meisten Patienten die Behandlung in Woche 12 abbrachen erster Besuch, bei dem Patienten mit einem CGI-S≥4 auch die Kriterien für „klinischen Non-Responder“ (CGI-S blieb gleich oder erhöht gegenüber dem Ausgangswert der Studie) erfüllen und für die Teilnahme an einer unverblindeten Verlängerungsstudie mit Atomoxetin infrage kommen könnten. Es gab Postmarketing-Berichte über Tics [siehe NEBENWIRKUNGEN ].
Angst bei Patienten mit ADHS und komorbiden Angststörungen
In zwei doppelblinden, placebokontrollierten Studien nach der Markteinführung wurde gezeigt, dass die Behandlung von Patienten mit ADHS und komorbiden Angststörungen mit STRATTERA 10 mg ihre Angstzustände nicht verschlimmert.
In einer 12-wöchigen doppelblinden, placebokontrollierten Studie wurden 176 Patienten im Alter von 8-17 Jahren, die die DSM-IV-Kriterien für ADHS erfüllten und mindestens eine der Angststörungen Trennungsangststörung, generalisierte Angststörung oder soziale Phobie waren, untersucht zufällig. Nach einer 2-wöchigen doppelblinden Placebo-Einleitung wurde mit STRATTERA 25 mg mit 0,8 mg/kg/Tag begonnen, mit Steigerung auf eine Zieldosis von 1,2 mg/kg/Tag (mediane Dosis 1,30 mg/kg/Tag +/-0,29 mg/kg/Tag). STRATTERA verschlimmerte die Angst bei diesen Patienten nicht, wie anhand der Pediatric Anxiety Rating Scale (PARS) bestimmt wurde. Von den 158 Patienten, die die doppelblinde Placebo-Lead-in-Studie beendeten, brachen 26 (16 %) Patienten die Studie ab.
In einer separaten 16-wöchigen, doppelblinden, placebokontrollierten Studie wurden 442 Patienten im Alter von 18 bis 65 Jahren, die die DSM-IV-Kriterien für ADHS und soziale Angststörung bei Erwachsenen erfüllten (von denen 23 % auch eine generalisierte Angststörung hatten), randomisiert. Nach einer 2-wöchigen doppelblinden Placebo-Einleitung wurde mit STRATTERA 10 mg mit 40 mg/Tag bis zu einer Höchstdosis von 100 mg/Tag begonnen (mittlere Tagesdosis 83 mg/Tag +/- 19,5 mg/Tag). STRATTERA 18 mg verschlimmerte die Angst bei diesen Patienten nicht, wie anhand der Liebowitz Social Anxiety Scale (LSAS) bestimmt wurde. Von den 413 Patienten, die die doppelblinde Placebo-Lead-in-Studie beendeten, brachen 149 (36,1 %) Patienten die Studie ab. Es gab Postmarketing-Berichte über Angstzustände [siehe NEBENWIRKUNGEN ] .
ÜBERDOSIS
Menschliche Erfahrung
Es liegen nur begrenzte Erfahrungen aus klinischen Studien mit einer Überdosierung von STRATTERA vor. Nach der Markteinführung wurden Todesfälle im Zusammenhang mit einer gemischten Einnahme einer Überdosis von STRATTERA 10 mg und mindestens einem anderen Arzneimittel gemeldet. Es gibt keine Berichte über Todesfälle im Zusammenhang mit einer Überdosierung von STRATTERA allein, einschließlich absichtlicher Überdosierung von Mengen bis zu 1400 mg. In einigen Fällen einer Überdosierung von STRATTERA wurde über Krampfanfälle berichtet. Die am häufigsten berichteten Symptome im Zusammenhang mit akuten und chronischen Überdosierungen von STRATTERA waren gastrointestinale Symptome, Somnolenz, Schwindel, Zittern und abnormales Verhalten. Hyperaktivität und Agitiertheit wurden ebenfalls berichtet. Anzeichen und Symptome, die mit einer leichten bis mittelschweren Aktivierung des sympathischen Nervensystems einhergehen (z. B. Tachykardie, erhöhter Blutdruck, Mydriasis, Mundtrockenheit), wurden ebenfalls beobachtet. Die meisten Ereignisse waren leicht bis mittelschwer. Weniger häufig gab es Berichte über QT-Verlängerung und mentale Veränderungen, einschließlich Orientierungslosigkeit und Halluzinationen [siehe KLINISCHE PHARMAKOLOGIE ].
Management einer Überdosierung
Wenden Sie sich an ein zertifiziertes Giftinformationszentrum, um aktuelle Anleitungen und Ratschläge zu erhalten. Da Atomoxetin stark an Proteine gebunden ist, ist eine Dialyse bei der Behandlung einer Überdosierung wahrscheinlich nicht sinnvoll.
KONTRAINDIKATIONEN
Überempfindlichkeit
STRATTERA ist kontraindiziert bei Patienten mit bekannter Überempfindlichkeit gegenüber Atomoxetin oder anderen Bestandteilen des Arzneimittels [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Monoaminoxidase-Hemmer (MAOI)
STRATTERA sollte nicht zusammen mit einem MAO-Hemmer oder innerhalb von 2 Wochen nach Absetzen eines MAO-Hemmers eingenommen werden. Die Behandlung mit einem MAO-Hemmer sollte nicht innerhalb von 2 Wochen nach Absetzen von STRATTERA begonnen werden. Bei anderen Arzneimitteln, die die Monoaminkonzentrationen im Gehirn beeinflussen, gab es Berichte über schwerwiegende, manchmal tödliche Reaktionen (einschließlich Hyperthermie, Starrheit, Myoklonus, autonome Instabilität mit möglichen schnellen Schwankungen der Vitalfunktionen und Veränderungen des Geisteszustands, einschließlich extremer Erregung bis hin zu Delirium und Koma), wenn es in Kombination mit einem MAOI eingenommen wird. Einige Fälle zeigten Merkmale, die dem malignen neuroleptischen Syndrom ähneln. Solche Reaktionen können auftreten, wenn diese Arzneimittel gleichzeitig oder in unmittelbarer Nähe gegeben werden [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Engwinkelglaukom
In klinischen Studien war die Anwendung von STRATTERA mit einem erhöhten Mydriasis-Risiko verbunden, weshalb die Anwendung bei Patienten mit Engwinkelglaukom nicht empfohlen wird.
Phäochromozytom
Schwerwiegende Reaktionen, einschließlich erhöhter Blutdruck und Tachyarrhythmie, wurden bei Patienten mit Phäochromozytom oder Phäochromozytom in der Vorgeschichte, die STRATTERA erhielten, berichtet. Daher sollte STRATTERA 18 mg nicht von Patienten mit Phäochromozytom oder Phäochromozytom in der Vorgeschichte eingenommen werden.
Schwere Herz-Kreislauf-Erkrankungen
STRATTERA 40 mg sollte nicht bei Patienten mit schweren Herz- oder Gefäßerkrankungen angewendet werden, deren Zustand sich voraussichtlich verschlechtern würde, wenn sie einen Anstieg des Blutdrucks oder der Herzfrequenz erfahren, der klinisch bedeutsam sein könnte (z Schläge pro Minute bei der Herzfrequenz). [Sehen WARNUNGEN UND VORSICHTSMASSNAHMEN ].
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Der genaue Mechanismus, durch den Atomoxetin seine therapeutischen Wirkungen bei der Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) entfaltet, ist unbekannt, es wird jedoch angenommen, dass er mit der selektiven Hemmung des präsynaptischen Noradrenalin-Transporters zusammenhängt, wie in Ex-vivo-Aufnahme- und Neurotransmitter-Depletionsstudien festgestellt wurde .
Pharmakodynamik
Eine Expositions-Wirkungs-Analyse, die Atomoxetin-Dosen (0,5, 1,2 oder 1,8 mg/kg/Tag) oder Placebo umfasste, zeigte, dass die Atomoxetin-Exposition mit der Wirksamkeit korreliert, gemessen anhand der Aufmerksamkeits-Defizit-/Hyperaktivitäts-Störungs-Bewertungsskala-IV-Elternversion: Vom Prüfarzt verabreicht und erzielt. Die Beziehung zwischen Exposition und Wirksamkeit war ähnlich der zwischen Dosis und Wirksamkeit beobachteten, wobei die medianen Expositionen bei den beiden höchsten Dosen zu nahezu maximalen Veränderungen gegenüber dem Ausgangswert führten [siehe Klinische Studien ].
Herzelektrophysiologie
Die Wirkung von STRATTERA auf die QTc-Verlängerung wurde in einer randomisierten, doppelblinden, positiven (Moxifloxacin 400 mg) und Placebo-kontrollierten Crossover-Studie an gesunden männlichen CYP2D6-Low-Metabolizern untersucht. Insgesamt 120 gesunden Probanden wurde STRATTERA (20 mg und 60 mg) zweimal täglich über 7 Tage verabreicht. In der Studie wurden keine großen Veränderungen des QTc-Intervalls (dh Zunahmen > 60 ms gegenüber dem Ausgangswert, absolutes QTc > 480 ms) beobachtet. Kleine Änderungen des QTc-Intervalls können jedoch nicht aus der aktuellen Studie ausgeschlossen werden, da die Studie die Assay-Sensitivität nicht nachweisen konnte. Es gab eine leichte Verlängerung des QTc-Intervalls mit erhöhter Atomoxetin-Konzentration.
Pharmakokinetik
Atomoxetin wird nach oraler Gabe gut resorbiert und durch Nahrung nur minimal beeinflusst. Es wird hauptsächlich durch oxidativen Metabolismus über den enzymatischen Cytochrom P450 2D6 (CYP2D6)-Weg und anschließende Glucuronidierung eliminiert. Atomoxetin hat eine Halbwertszeit von etwa 5 Stunden. Ein Bruchteil der Bevölkerung (etwa 7 % der Kaukasier und 2 % der Afroamerikaner) sind schlechte Metabolisierer (PMs) von CYP2D6-metabolisierten Arzneimitteln. Diese Personen haben eine verringerte Aktivität auf diesem Weg, was zu 10-fach höheren AUCs, 5-fach höheren maximalen Plasmakonzentrationen und einer langsameren Elimination (Plasmahalbwertszeit von etwa 24 Stunden) von Atomoxetin im Vergleich zu Personen mit normaler Aktivität führt [extensive metabolizers (EMs )]. Medikamente, die CYP2D6 hemmen, wie Fluoxetin, Paroxetin und Chinidin, verursachen ähnliche Erhöhungen der Exposition.
Die Pharmakokinetik von Atomoxetin wurde bei mehr als 400 Kindern und Jugendlichen in ausgewählten klinischen Studien untersucht, hauptsächlich unter Verwendung von populationspharmakokinetischen Studien. Einzelne pharmakokinetische Daten zur Einzeldosis und zum Steady-State wurden auch bei Kindern, Jugendlichen und Erwachsenen erhoben. Wenn die Dosen auf eine mg/kg-Basis normalisiert wurden, wurden bei Kindern, Jugendlichen und Erwachsenen ähnliche Halbwertszeit-, Cmax- und AUC-Werte beobachtet. Clearance und Verteilungsvolumen nach Anpassung an das Körpergewicht waren ebenfalls ähnlich.
Absorption und Verteilung
Atomoxetin wird nach oraler Verabreichung schnell resorbiert, mit einer absoluten Bioverfügbarkeit von etwa 63 % bei EMs und 94 % bei PMs. Maximale Plasmakonzentrationen (Cmax) werden etwa 1 bis 2 Stunden nach der Einnahme erreicht.
STRATTERA 18 mg kann mit oder ohne Nahrung verabreicht werden. Die Verabreichung von STRATTERA zusammen mit einer fettreichen Standardmahlzeit bei Erwachsenen hatte keinen Einfluss auf das Ausmaß der oralen Resorption von Atomoxetin (AUC), verringerte jedoch die Resorptionsrate, was zu einer um 37 % niedrigeren Cmax und einer um 3 Stunden verzögerten Tmax führte. In klinischen Studien mit Kindern und Jugendlichen führte die Einnahme von STRATTERA zusammen mit Nahrung zu einer um 9 % niedrigeren Cmax.
Das Verteilungsvolumen im Steady State nach intravenöser Verabreichung beträgt 0,85 l/kg, was darauf hinweist, dass sich Atomoxetin hauptsächlich im gesamten Körperwasser verteilt. Das Verteilungsvolumen ist nach Normalisierung auf das Körpergewicht über den Gewichtsbereich des Patienten ähnlich.
Bei therapeutischen Konzentrationen sind 98 % von Atomoxetin im Plasma an Protein, hauptsächlich Albumin, gebunden.
Stoffwechsel und Ausscheidung
Atomoxetin wird hauptsächlich über den enzymatischen Weg CYP2D6 metabolisiert. Menschen mit reduzierter Aktivität in diesem Signalweg (PMs) haben höhere Plasmakonzentrationen von Atomoxetin im Vergleich zu Menschen mit normaler Aktivität (EMs). Bei PMs ist die AUC von Atomoxetin etwa 10-mal größer und Css,max etwa 5-mal größer als bei EMs. Labortests sind verfügbar, um CYP2D6-PMs zu identifizieren. Die gleichzeitige Anwendung von STRATTERA mit potenten CYP2D6-Inhibitoren wie Fluoxetin, Paroxetin oder Chinidin führt zu einem erheblichen Anstieg der Atomoxetin-Plasmaexposition und eine Dosisanpassung kann erforderlich sein [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Atomoxetin hemmte oder induziert den CYP2D6-Signalweg nicht.
Der wichtigste oxidative Metabolit, der unabhängig vom CYP2D6-Status gebildet wird, ist 4-Hydroxyatomoxetin, das glucuronidiert ist. 4-Hydroxyatomoxetin ist als Inhibitor des Norepinephrin-Transporters gleich wirksam wie Atomoxetin, zirkuliert jedoch im Plasma in viel geringeren Konzentrationen (1 % der Atomoxetin-Konzentration in EMs und 0,1 % der Atomoxetin-Konzentration in PMs). 4-Hydroxyatomoxetin wird hauptsächlich von CYP2D6 gebildet, aber bei PMs wird 4-Hydroxyatomoxetin langsamer von mehreren anderen Cytochrom-P450-Enzymen gebildet. N-Desmethylatomoxetin wird von CYP2C19 und anderen Cytochrom-P450-Enzymen gebildet, hat aber im Vergleich zu Atomoxetin eine wesentlich geringere pharmakologische Aktivität und zirkuliert im Plasma in geringeren Konzentrationen (5 % der Atomoxetin-Konzentration in EMs und 45 % der Atomoxetin-Konzentration in PMs).
Die mittlere scheinbare Plasma-Clearance von Atomoxetin nach oraler Gabe bei erwachsenen EM beträgt 0,35 l/h/kg und die mittlere Halbwertszeit 5,2 Stunden. Nach oraler Verabreichung von Atomoxetin an PMs beträgt die mittlere scheinbare Plasmaclearance 0,03 l/h/kg und die mittlere Halbwertszeit 21,6 Stunden. Bei PMs ist die AUC von Atomoxetin etwa 10-mal größer und Css,max etwa 5-mal größer als bei EMs. Die Eliminationshalbwertszeit von 4-Hydroxyatomoxetin ist bei EM-Patienten ähnlich der von N-Desmethylatomoxetin (6 bis 8 Stunden), während die Halbwertszeit von N-Desmethylatomoxetin bei PM-Patienten viel länger ist (34 bis 40 Stunden).
Atomoxetin wird hauptsächlich als 4-Hydroxyatomoxetin-O-glucuronid ausgeschieden, hauptsächlich im Urin (mehr als 80 % der Dosis) und in geringerem Maße im Stuhl (weniger als 17 % der Dosis). Nur ein kleiner Teil der 40-mg-Dosis von STRATTERA wird als unverändertes Atomoxetin ausgeschieden (weniger als 3 % der Dosis), was auf eine umfangreiche Biotransformation hinweist.
[Sehen Verwendung in bestimmten Bevölkerungsgruppen ].
Klinische Studien
ADHS-Studien bei Kindern und Jugendlichen
Akutstudien
Die Wirksamkeit von STRATTERA 40 mg bei der Behandlung von ADHS wurde in 4 randomisierten, doppelblinden, placebokontrollierten Studien mit pädiatrischen Patienten (im Alter von 6 bis 18 Jahren) nachgewiesen. Ungefähr ein Drittel der Patienten erfüllte die DSM-IV-Kriterien für den unaufmerksamen Subtyp und zwei Drittel erfüllten die Kriterien sowohl für den unaufmerksamen als auch den hyperaktiven/impulsiven Subtyp.
Anzeichen und Symptome von ADHS wurden durch einen Vergleich der mittleren Veränderung vom Ausgangswert bis zum Endpunkt für mit STRATTERA und Placebo behandelte Patienten unter Verwendung einer Intent-to-treat-Analyse des primären Zielparameters, der vom Prüfarzt verabreichten und bewerteten ADHS-Bewertungsskala-IV- bewertet. Gesamtpunktzahl der Elternversion (ADHDRS), einschließlich hyperaktiver/impulsiver und unaufmerksamer Subskalen. Jedes Element auf ADHDRS wird direkt einem Symptomkriterium für ADHS im DSM-IV zugeordnet.
In Studie 1, einer 8-wöchigen randomisierten, doppelblinden, placebokontrollierten Dosis-Wirkungs-Akutbehandlungsstudie mit Kindern und Jugendlichen im Alter von 8 bis 18 Jahren (N = 297), erhielten die Patienten entweder eine feste Dosis STRATTERA (0,5, 1,2 oder 1,8 mg/kg/Tag) oder Placebo. STRATTERA 10 mg wurde als aufgeteilte Dosis am frühen Morgen und am späten Nachmittag/frühen Abend verabreicht. Bei den 2 höheren Dosen waren die Verbesserungen der ADHS-Symptome bei den mit STRATTERA behandelten Patienten statistisch signifikant besser als bei den mit Placebo behandelten Patienten, gemessen anhand der ADHDRS-Skala. Die Dosis von 1,8 mg/kg/Tag STRATTERA 25 mg brachte keinen zusätzlichen Nutzen gegenüber der Dosis von 1,2 mg/kg/Tag. Die Dosis von 0,5 mg/kg/Tag STRATTERA war Placebo nicht überlegen.
In Studie 2, einer 6-wöchigen randomisierten, doppelblinden, placebokontrollierten Akutbehandlungsstudie mit Kindern und Jugendlichen im Alter von 6 bis 16 Jahren (N = 171), erhielten die Patienten entweder STRATTERA 40 mg oder Placebo. STRATTERA wurde als Einzeldosis am frühen Morgen verabreicht und entsprechend dem klinischen Ansprechen gewichtsangepasst bis zu einer Höchstdosis von 1,5 mg/kg/Tag titriert. Die mittlere Enddosis von STRATTERA 10 mg betrug ungefähr 1,3 mg/kg/Tag. ADHS-Symptome wurden unter STRATTERA im Vergleich zu Placebo statistisch signifikant gebessert, gemessen anhand der ADHDRS-Skala. Diese Studie zeigt, dass STRATTERA wirksam ist, wenn es einmal täglich morgens verabreicht wird.
In zwei identischen, 9-wöchigen, akuten, randomisierten, doppelblinden, placebokontrollierten Studien mit Kindern im Alter von 7 bis 13 Jahren (Studie 3, N = 147; Studie 4, N = 144) wurden STRATTERA 40 mg und Methylphenidat mit Placebo verglichen . STRATTERA 18 mg wurde als aufgeteilte Dosis am frühen Morgen und am späten Nachmittag (nach der Schule) verabreicht und entsprechend dem klinischen Ansprechen gewichtsangepasst titriert. Die maximal empfohlene STRATTERA-Dosis betrug 2,0 mg/kg/Tag. Die mittlere Enddosis von STRATTERA 18 mg betrug in beiden Studien etwa 1,6 mg/kg/Tag. In beiden Studien verbesserten sich die ADHS-Symptome unter STRATTERA 10 mg statistisch signifikant stärker als unter Placebo, gemessen auf der ADHDRS-Skala.
Die Untersuchung von Bevölkerungsuntergruppen basierend auf Geschlecht und Alter (
Wartungsstudie
Die Wirksamkeit von STRATTERA 18 mg bei der Erhaltungstherapie von ADHS wurde in einer ambulanten Studie mit Kindern und Jugendlichen (Alter 6-15 Jahre) nachgewiesen. Patienten, die die DSM-IV-Kriterien für ADHS erfüllten und während einer anfänglichen 10-wöchigen offenen Behandlungsphase mit STRATTERA (1,2 bis 1,8 mg/kg/Tag) über etwa 4 Wochen ein kontinuierliches Ansprechen zeigten, wurden randomisiert auf die Fortsetzung ihrer aktuellen Dosis von STRATTERA (N =292) oder Placebo (N=124) unter doppelblinder Behandlung zur Beobachtung eines Rückfalls. Das Ansprechen während der Open-Label-Phase wurde definiert als CGI-ADHD-S-Score ≤ 2 und eine Reduktion des ADHDRS-IV-Parent:Inv-Gesamtscores um mindestens 25 % gegenüber dem Ausgangswert. Patienten, die STRATTERA zugewiesen wurden und während der ersten doppelblinden Behandlungsphase für etwa 8 Monate ein kontinuierliches Ansprechen zeigten, wurden erneut randomisiert und erhielten doppelblind die Fortsetzung ihrer aktuellen STRATTERA-Dosis (N = 81) oder Placebo (N = 82). Behandlung zur Beobachtung eines Rückfalls. Ein Rückfall während der doppelblinden Phase wurde als Anstieg des CGI-ADHD-S-Scores um mindestens 2 ab dem Ende der Open-Label-Phase definiert und der ADHDRS-IV-Parent:Inv-Gesamtscore kehrte für 2 auf ≥ 90 % des Studieneintrittsscores zurück aufeinanderfolgende Besuche. In beiden doppelblinden Phasen traten bei Patienten, die die STRATTERA-Behandlung fortsetzten, signifikant längere Zeiten bis zum Rückfall auf als bei Patienten, die Placebo erhielten.
ADHS-Studien bei Erwachsenen
Die Wirksamkeit von STRATTERA bei der Behandlung von ADHS wurde in 2 randomisierten, doppelblinden, placebokontrollierten klinischen Studien mit erwachsenen Patienten ab 18 Jahren nachgewiesen, die die DSM-IV-Kriterien für ADHS erfüllten.
Anzeichen und Symptome von ADHS wurden anhand der vom Prüfarzt verabreichten Conners Adult ADHD Rating Scale Screening Version (CAARS), einer 30-Punkte-Skala, bewertet. Der primäre Wirksamkeitsmaßstab war der 18-Punkte-Gesamt-ADHS-Symptom-Score (die Summe der Unterskalen für Unaufmerksamkeit und Hyperaktivität/Impulsivität aus dem CAARS), der durch einen Vergleich der mittleren Veränderung vom Ausgangswert zum Endpunkt unter Verwendung einer Intent-to-treat-Analyse bewertet wurde.
In 2 identischen, 10-wöchigen, randomisierten, doppelblinden, placebokontrollierten Akutbehandlungsstudien (Studie 5, N = 280; Studie 6, N = 256) erhielten die Patienten entweder STRATTERA 25 mg oder Placebo. STRATTERA wurde als aufgeteilte Dosis am frühen Morgen und am späten Nachmittag/frühen Abend verabreicht und entsprechend dem klinischen Ansprechen in einem Bereich von 60 bis 120 mg/Tag titriert. Die mittlere Enddosis von STRATTERA 40 mg betrug in beiden Studien etwa 95 mg/Tag. In beiden Studien verbesserten sich die ADHS-Symptome unter STRATTERA statistisch signifikant, gemessen am ADHS-Symptom-Score der CAARS-Skala.
Die Untersuchung von Bevölkerungsuntergruppen basierend auf Geschlecht und Alter (
INFORMATIONEN ZUM PATIENTEN
STRATTERA® (Stra-TAIR-a) (Atomoxetin) Kapseln
Lesen Sie den mit STRATTERA® gelieferten Medikationsleitfaden, bevor Sie oder Ihr Kind mit der Einnahme beginnen und jedes Mal, wenn Sie eine Nachfüllung erhalten. Möglicherweise gibt es neue Informationen. Dieser Medikationsleitfaden ersetzt nicht das Gespräch mit Ihrem Arzt über Ihre Behandlung oder die Behandlung Ihres Kindes mit STRATTERA.
Was sind die wichtigsten Informationen, die ich über STRATTERA 18 mg wissen sollte?
Folgendes wurde bei der Anwendung von STRATTERA berichtet:
Kinder und Jugendliche denken manchmal an Selbstmord, und viele berichten, dass sie versucht haben, sich das Leben zu nehmen. Ergebnisse klinischer Studien zu STRATTERA mit über 2200 ADHS-Patienten im Kindes- und Jugendalter deuten darauf hin, dass bei manchen Kindern und Jugendlichen ein höheres Risiko für Selbstmordgedanken oder -handlungen besteht. Obwohl in diesen Studien keine Suizide auftraten, entwickelten 4 von 1000 Patienten Suizidgedanken. Informieren Sie den Arzt Ihres Kindes oder Jugendlichen, wenn bei Ihrem Kind oder Jugendlichen (oder in der Familienanamnese):
Die Wahrscheinlichkeit für Selbstmordgedanken und -handlungen kann höher sein:
Verhindern Sie Suizidgedanken und -handlungen bei Ihrem Kind oder Teenager, indem Sie:
Achten Sie während der Behandlung mit STRATTERA 25 mg auf die folgenden Anzeichen bei Ihrem Kind oder Teenager:
Rufen Sie sofort den Arzt Ihres Kindes oder Teenagers an, wenn eines der oben genannten Anzeichen auftritt, insbesondere wenn es neu, plötzlich oder schwerwiegend ist. Ihr Kind oder Teenager muss möglicherweise engmaschig auf Selbstmordgedanken und -handlungen überwacht werden oder es muss eine Änderung der Medizin vorgenommen werden.
STRATTERA kann bei manchen Patienten Leberschäden verursachen. Rufen Sie sofort Ihren Arzt an, wenn Sie oder Ihr Kind die folgenden Anzeichen von Leberproblemen haben:
Informieren Sie Ihren Arzt, wenn Sie oder Ihr Kind Herzprobleme, Herzfehler, Bluthochdruck oder eine Familienanamnese dieser Probleme haben. Ihr Arzt sollte Sie oder Ihr Kind vor Beginn der Behandlung mit STRATTERA sorgfältig auf Herzprobleme untersuchen.
Ihr Arzt sollte Ihren Blutdruck oder den Blutdruck und die Herzfrequenz Ihres Kindes während der Behandlung mit STRATTERA regelmäßig kontrollieren.
Rufen Sie sofort Ihren Arzt an, wenn bei Ihnen oder Ihrem Kind während der Einnahme von STRATTERA Anzeichen von Herzproblemen wie Brustschmerzen, Kurzatmigkeit oder Ohnmacht auftreten.
Rufen Sie bei neuen psychischen Symptomen sofort den Arzt Ihres Kindes oder Jugendlichen an da eine Anpassung oder Beendigung der Behandlung mit STRATTERA 40 mg möglicherweise in Betracht gezogen werden muss.
Was ist STRATTERA 18 mg?
STRATTERA ist ein selektiver Noradrenalin-Wiederaufnahmehemmer. Es wird zur Behandlung der Aufmerksamkeitsdefizit- und Hyperaktivitätsstörung (ADHS) eingesetzt. STRATTERA kann helfen, die Aufmerksamkeit zu erhöhen und Impulsivität und Hyperaktivität bei Patienten mit ADHS zu verringern.
STRATTERA 10 mg sollte als Teil eines Gesamtbehandlungsprogramms für ADHS verwendet werden, das Beratung oder andere Therapien umfassen kann.
STRATTERA wurde bei Kindern unter 6 Jahren nicht untersucht.
Wer sollte STRATTERA nicht einnehmen?
STRATTERA darf nicht eingenommen werden, wenn Sie oder Ihr Kind:
STRATTERA ist möglicherweise nicht das Richtige für Sie oder Ihr Kind. Bevor Sie mit STRATTERA beginnen, informieren Sie Ihren Arzt oder den Arzt Ihres Kindes über alle Gesundheitszustände (oder eine Familienanamnese), einschließlich:
Kann STRATTERA mit anderen Arzneimitteln eingenommen werden?
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie oder Ihr Kind einnehmen, einschließlich verschreibungspflichtiger und nicht verschreibungspflichtiger Arzneimittel, Vitamine und pflanzlicher Nahrungsergänzungsmittel. STRATTERA und einige Arzneimittel können miteinander interagieren und schwerwiegende Nebenwirkungen verursachen. Ihr Arzt wird entscheiden, ob STRATTERA zusammen mit anderen Arzneimitteln eingenommen werden kann.
Informieren Sie Ihren Arzt insbesondere, wenn Sie oder Ihr Kind Folgendes einnehmen:
Informieren Sie sich über die Arzneimittel, die Sie oder Ihr Kind einnehmen. Führen Sie eine Liste Ihrer Arzneimittel mit sich, um sie Ihrem Arzt und Apotheker zu zeigen.
Beginnen Sie während der Einnahme von STRATTERA mit keinem neuen Arzneimittel, ohne vorher mit Ihrem Arzt gesprochen zu haben.
Wie sollte STRATTERA eingenommen werden?
Welche Nebenwirkungen kann STRATTERA haben?
Sehen „Was sind die wichtigsten Informationen, die ich über STRATTERA 10 mg wissen sollte?“ Informationen zu gemeldeten Selbstmordgedanken und -handlungen, anderen psychischen Problemen, schweren Leberschäden und Herzproblemen.
Andere schwerwiegende Nebenwirkungen sind:
Häufige Nebenwirkungen bei Kindern und Jugendlichen sind:
Häufige Nebenwirkungen bei Erwachsenen sind:
Weitere Informationen für Kinder, Jugendliche und Erwachsene:
Dies ist keine vollständige Liste möglicher Nebenwirkungen. Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist STRATTERA aufzubewahren?
Allgemeine Informationen zu STRATTERA
Arzneimittel werden manchmal für andere als die in einem Arzneimittelleitfaden aufgeführten Zwecke verschrieben. Verwenden Sie STRATTERA 25 mg nicht für einen Zustand, für den es nicht verschrieben wurde. Geben Sie STRATTERA nicht an andere Personen, selbst wenn sie die gleiche Erkrankung haben. Es kann ihnen schaden.
Dieser Arzneimittelleitfaden fasst die wichtigsten Informationen zu STRATTERA zusammen. Wenn Sie weitere Informationen wünschen, sprechen Sie mit Ihrem Arzt. Sie können Ihren Arzt oder Apotheker um Informationen über STRATTERA bitten, die für medizinisches Fachpersonal geschrieben wurden. Weitere Informationen zu STRATTERA erhalten Sie telefonisch unter 1-800-Lilly-Rx (1-800-545-5979) oder unter www.strattera.com.
Was sind die Inhaltsstoffe von STRATTERA 40 mg?
Wirkstoff: Atomoxetinhydrochlorid.
Inaktive Zutaten: vorgelatinierte Stärke, Dimethicon, Gelatine, Natriumlaurylsulfat, FD&CBlue Nr. 2, synthetisches gelbes Eisenoxid, Titandioxid, rotes Eisenoxid und essbare schwarze Tinte.
Dieser Medikationsleitfaden wurde von der US Food and Drug Administration genehmigt.
Was ist Sumycin und wie wird es angewendet?
Sumycin (Tetracyclinhydrochlorid) ist ein Antibiotikum, das zur Behandlung vieler verschiedener bakterieller Infektionen wie Harnwegsinfektionen, Akne, Gonorrhoe, Chlamydien und anderen verwendet wird. Sumycin ist in generischer Form erhältlich.
Welche Nebenwirkungen hat Sumycin 250mg?
Häufige Nebenwirkungen von Sumycin sind:
Um die Entwicklung arzneimittelresistenter Bakterien zu reduzieren und die Wirksamkeit von Sumycin '250' und Sumycin '500' Tabletten (Tetracyclin-Hydrochlorid-Tabletten) und andere antibakterielle Arzneimittel, Sumycin '250' und Sumycin '500' Tabletten (Tetracyclinhydrochlorid-Tabletten) sollten nur zur Behandlung oder Vorbeugung von Infektionen angewendet werden, die nachweislich oder mit starkem Verdacht auf Bakterien zurückzuführen sind.
BEZEICHNUNG
Sumycin zur oralen Verabreichung enthält Tetracyclin, ein aus Streptomyces aureofaciens isoliertes Antibiotikum. Tetracyclin wird chemisch als 4-(Dimethylamino)-1, 4, 4a, 5, 5a, 6, 11, 12a-octa-hydro-3, 6, 10, 12, 12a-pentahydroxy-6-methyl-1, 11 beschrieben -dioxo-2-napthacencarboxamid; seine Strukturformel lautet:
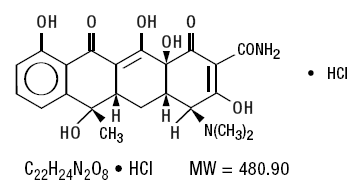
Sumycin '250' und Sumycin '500' Tabletten (Tetracyclin-Hydrochlorid-Tabletten) sind zur oralen Verabreichung als Tabletten mit 250 mg bzw. 500 mg Tetracyclin-Hydrochlorid erhältlich. Inaktive Inhaltsstoffe: Farbstoffe (D&C Red Nr. 30 Aluminiumlack, Titandioxid), Hypromellose, wasserfreie Lactose, Magnesiumstearat, mikrokristalline Cellulose, Povidon, vorgelatinierte Stärke, Stearinsäure. Darüber hinaus enthalten 250 mg Methylenchlorid, Hydroxypropylcellulose, Triacetin und 500 mg enthalten Polyethylenglykol, Polyparaben, Methylparaben, Natriumcitrat, Kaliumsorbat, Propylparaben und Xanthangummi.
INDIKATIONEN
Um die Entwicklung arzneimittelresistenter Bakterien zu reduzieren und die Wirksamkeit von Sumycin '250' und Sumycin '500' Tabletten (Tetracyclin-Hydrochlorid-Tabletten) und anderen antibakteriellen Arzneimitteln aufrechtzuerhalten, Sumycin '250' und Sumycin '500' Tabletten (Tetracyclin-Hydrochlorid-Tabletten). ) sollte nur zur Behandlung oder Vorbeugung von Infektionen angewendet werden, die nachweislich oder mit starkem Verdacht auf empfindliche Bakterien zurückzuführen sind. Wenn Kultur- und Empfindlichkeitsinformationen verfügbar sind, sollten diese bei der Auswahl oder Änderung der antibakteriellen Therapie berücksichtigt werden. In Ermangelung solcher Daten können lokale Epidemiologie und Anfälligkeitsmuster zur empirischen Auswahl der Therapie beitragen.
Tetracyclinhydrochlorid ist angezeigt zur Behandlung der folgenden Infektionen:
Rocky-Mountain-Fleckfieber, Typhusfieber und die Typhusgruppe, Q-Fieber, Rickettsienpocken und durch Rickettsien verursachtes Zeckenfieber.
Durch Mycoplasma pneumoniae verursachte Atemwegsinfektionen
Lymphogranuloma venereum, verursacht durch Chlamydia trachomatis
Psittakose und Ornithose durch Chlamydia psittaci
Durch Chlamydia trachomatis verursachtes Trachom, obwohl der Infektionserreger laut Immunfluoreszenz nicht immer eliminiert wird
Einschlusskonjunktivitis verursacht durch Chlamydia trachomatis
Tetracyclinhydrochlorid ist indiziert zur Behandlung von durch Chlamydia trachomatis verursachten unkomplizierten urethralen, endozervikalen oder rektalen Infektionen bei Erwachsenen
Nichtgonokokken-Urethritis verursacht durch Ureaplasma urealyticum Rückfallfieber durch Borrelia recurrentis
Tetracyclinhydrochlorid ist auch angezeigt zur Behandlung von Infektionen, die durch die folgenden gramnegativen Mikroorganismen verursacht werden:
Schanker, verursacht durch Haemophilus ducreyi
Pest durch Yersinia pestis (früher Pasteurella pestis)
Tularämie durch Francisella tularensis (früher Pasteurella tularensis)
Cholera verursacht durch Vibrio cholerae (früher Vibrio comma)
Campylobacter fetus-Infektionen, verursacht durch Campylobacter fetus (früher Vibrio fetus)
Brucellose durch Brucella-Spezies (in Verbindung mit Streptomycin)
Bartonellose durch Bartonella bacilliformis
Granuloma inguinale, verursacht durch Calymmatobacterium granulomatis
Da sich viele Stämme der folgenden Gruppen von Mikroorganismen als resistent gegen Tetracyclinhydrochlorid erwiesen haben, werden Kultur- und Empfindlichkeitstests empfohlen.
Tetracyclinhydrochlorid ist indiziert zur Behandlung von Infektionen, die durch die folgenden gramnegativen Mikroorganismen verursacht werden, wenn bakteriologische Tests eine angemessene Empfindlichkeit gegenüber dem Arzneimittel anzeigen:
Escherichia coli
Enterobacter aerogenes (früher Aerobacter aerogenes)
Shigella-Arten
Acinetobacter-Arten [früher Mima-Arten und Herellea-Arten]
Durch Haemophilus influenzae verursachte Atemwegsinfektionen
Atemwegs- und Harnwegsinfektionen, verursacht durch Klebsiella-Arten
Tetracyclinhydrochlorid ist indiziert zur Behandlung von Infektionen, die durch die folgenden grampositiven Mikroorganismen verursacht werden, wenn bakteriologische Tests eine entsprechende Empfindlichkeit gegenüber dem Arzneimittel ergeben haben:
Bei Infektionen der oberen Atemwege, verursacht durch Streptococcus pneumoniae (früher Diplococcus pneumoniae)
Haut- und Hautstrukturinfektionen, die durch Staphylococcus aureus verursacht werden. Tetracycline sind nicht die Mittel der Wahl bei der Behandlung jeglicher Art von Staphylokokkeninfektionen
Wenn Penicillin kontraindiziert ist, ist Tetracyclinhydrochlorid ein alternatives Medikament zur Behandlung der folgenden Infektionen:
Unkomplizierter Tripper, verursacht durch Neisseria gonorrhoeae
Syphilis verursacht durch Treponema pallidum
Gieren verursacht durch Treponema pertenue
Listeriose durch Listeria monocytogenes
Milzbrand durch Bacillus anthracis
Vincent-Infektion, verursacht durch Fusobacterium fusiforme
Aktinomykose, verursacht durch Actinomyces israelii
Infektionen durch Clostridia-Arten
Bei akuter intestinaler Amöbiasis können die Tetracyclin-Hydrochloride eine nützliche Zusatztherapie zu Amebiziden sein.
Bei schwerer Akne können die Tetracyclinhydrochloride eine sinnvolle Zusatztherapie sein.
DOSIERUNG UND ANWENDUNG
Erwachsene: Die übliche Tagesdosis beträgt 1 bis 2 g; bei leichten bis mittelschweren Infektionen: 500 mg 2-mal täglich oder 250 mg 4-mal täglich; bei schweren Infektionen können höhere Dosierungen wie 500 mg 4-mal täglich erforderlich sein.
Für Kinder über acht Jahren: Die übliche Tagesdosis beträgt 10 bis 20 mg/lb (25 bis 50 mg/kg) Körpergewicht, aufgeteilt in vier gleiche Dosen.
Die Therapie sollte mindestens 24 bis 48 Stunden nach Abklingen der Symptome und des Fiebers fortgesetzt werden.
Die Behandlung von Brucellose, 500 mg Tetracyclin viermal täglich für drei Wochen, sollte begleitet werden von Streptomycin, 1 g intramuskulär zweimal täglich in der ersten Woche und einmal täglich in der zweiten Woche.
Zur Behandlung von unkomplizierter Gonorrhoe sieben Tage lang alle sechs Stunden 500 mg.
Zur Behandlung der Syphilis sollten insgesamt 30 bis 40 g in gleichmäßig verteilten Dosen über einen Zeitraum von 10 bis 15 Tagen verabreicht werden. Eine engmaschige Nachsorge, einschließlich Labortests, wird empfohlen.
Unkomplizierte urethrale, endozervikale oder rektale Infektion bei Erwachsenen, verursacht durch Chlamydia trachomatis: 500 mg oral, viermal täglich für mindestens sieben Tage.
Bei schwerer Akne, die nach Einschätzung des Arztes eine Langzeitbehandlung erfordert, beträgt die empfohlene Anfangsdosis 1 g täglich in aufgeteilten Dosen. Wenn eine Besserung festgestellt wird, normalerweise innerhalb einer Woche, sollte die Dosierung schrittweise auf Erhaltungsspiegel im Bereich von 125 bis 500 mg täglich reduziert werden. Bei manchen Patienten kann es möglich sein, eine adäquate Remission der Läsionen mit alternierender oder intermittierender Therapie aufrechtzuerhalten. Die Tetracyclin-Therapie von Akne sollte die anderen bekannten Standardmaßnahmen ergänzen.
Bei Patienten mit eingeschränkter Nierenfunktion (vgl WARNUNGEN sollte die Gesamtdosis verringert werden, indem die empfohlenen Einzeldosen reduziert und/oder die Zeitabstände zwischen den Dosen verlängert werden.
Bei der Behandlung von Streptokokkeninfektionen sollte eine therapeutische Dosis Tetracyclin für mindestens 10 Tage verabreicht werden.
Begleittherapie: Die Resorption von Tetracyclinen wird durch Antazida, die Aluminium, Calcium oder Magnesium enthalten, und eisenhaltige Präparate beeinträchtigt.
Lebensmittel und einige Milchprodukte beeinträchtigen die Absorption ebenfalls.
Die Verabreichung ausreichender Flüssigkeitsmengen zusammen mit der Tablette und insbesondere Kapselformulierungen von Tetracyclin wird empfohlen, um das Medikament herunterzuspülen und das Risiko einer Reizung und Ulzeration der Speiseröhre zu verringern (siehe Abschnitt 4.4). NEBENWIRKUNGEN )
WIE GELIEFERT
Sumycin® Tabletten (Tetracyclin-Hydrochlorid-Tabletten USP)
Lagerung
Bewahren Sie die Tabletten bei Raumtemperatur auf; Vermeiden Sie übermäßige Hitze. In dichte, lichtbeständige Behälter füllen.
Hergestellt von: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA. Hergestellt für: Par Pharmaceutical, Inc. Spring Valley, NY 10977. USA. Überarbeitet 03/04. Revisionsdatum der FDA: 22.07.1997
NEBENWIRKUNGEN
Magen-Darm: Anorexie, epigastrische Beschwerden, Übelkeit, Erbrechen, Durchfall, massiger weicher Stuhl, Stomatitis, Halsschmerzen, Glossitis, schwarze Haarzunge, Dysphagie, Heiserkeit, Enterokolitis und entzündliche Läsionen (mit Candida-Wucherung) im Anogenitalbereich, einschließlich Proktitis und Pruritus ani . Seltene Fälle von Ösophagitis und Ösophagusulzerationen wurden bei Patienten berichtet, die insbesondere die Kapsel- und auch die Tablettenform von Tetracyclinen erhielten. Es wurde berichtet, dass die meisten Patienten unmittelbar vor dem Schlafengehen Medikamente einnahmen (siehe DOSIERUNG UND ANWENDUNG ). Diese Reaktionen wurden sowohl durch die orale als auch durch die parenterale Verabreichung von Tetracyclinen verursacht, treten aber nach parenteraler Anwendung weniger häufig auf.
Haut und Hautstrukturen: makulopapulöse und erythematöse Hautausschläge. Exfoliative Dermatitis wurde berichtet, ist aber selten. Onycholyse und Verfärbung der Nägel wurden selten berichtet. Lichtempfindlichkeit ist aufgetreten. (Sehen WARNUNGEN ).
Nierentoxizität: Anstiege von BUN wurden berichtet und sind offensichtlich dosisabhängig. (Sehen WARNUNGEN. )
Hepatische Cholestase: wurde selten berichtet und ist normalerweise mit hohen Dosierungen von Tetracyclin verbunden.
Überempfindlichkeitsreaktionen: Anaphylaxie; serumkrankheitsähnliche Reaktionen wie Fieber, Hautausschlag und Arthralgie; Urtikaria, angioneurotisches Ödem, anaphylaktoide Purpura, Perikarditis, Exazerbation von systemischem Lupus erythematodes.
Hämatologisch: Blut: Anämie, hämolytische Anämie, Thrombozytopenie, thrombozytopenische Purpura, Neutropenie und Eosinophilie wurden berichtet.
Sonstig: Schwindel und Kopfschmerzen wurden berichtet.
Es wurde berichtet, dass Tetracycline, wenn sie über einen längeren Zeitraum verabreicht werden, eine braun-schwarze mikroskopische Verfärbung der Schilddrüse hervorrufen. Es sind keine Anomalien der Schilddrüsenfunktion bekannt. Ausgewölbte Fontanellen bei Säuglingen und intrakranielle Hypertonie bei Erwachsenen wurden berichtet. (Sehen VORSICHTSMASSNAHMEN-Allgemein. )
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
PENICILLIN - Da bakteriostatische Arzneimittel wie Tetracyclin die bakterizide Wirkung von Penicillin beeinträchtigen können, ist es ratsam, die Verabreichung von Tetracyclin in Verbindung mit Penicillin zu vermeiden.
ANTIKOAGULANZIEN - Da gezeigt wurde, dass die Tetracycline die Plasma-Prothrombin-Aktivität senken, kann es bei Patienten, die eine Antikoagulanzien-Therapie erhalten, erforderlich sein, ihre Antikoagulans-Dosierung nach unten anzupassen.
ANTAZIDIEN UND EISENHALTIGE PRODUKTE - Die Resorption von Tetracyclin wird durch Antazida, die Aluminium, Calcium oder Magnesium enthalten, und eisenhaltige Präparate beeinträchtigt.
ORALE VERHÜTUNGSMITTEL -Die gleichzeitige Anwendung von Tetracyclin kann orale Kontrazeptiva weniger wirksam machen.
METHOXYFLURAN - Es wurde berichtet, dass die gleichzeitige Anwendung von Tetracyclin und Methoxyfluran zu tödlicher Nierentoxizität führt.
WARNUNGEN
ANTIBIOTIKA DER TETRACYCLIN-KLASSE KÖNNEN FÖTALEN SCHÄDEN VERURSACHEN, WENN SIE EINER SCHWANGEREN FRAU VERABREICHT WERDEN. WENN EIN TETRACYCLIN WÄHREND DER SCHWANGERSCHAFT ANGENOMMEN WIRD ODER DIE PATIENTIN WÄHREND DER EINNAHME DIESER ARZNEIMITTEL SCHWANGER WIRD, SOLLTE DIE PATIENTIN ÜBER DIE POTENZIELLE GEFAHR FÜR DEN FÖTUS INFORMIERT WERDEN.
DIE ANWENDUNG VON ARZNEIMITTELN DER TETRACYCLINE-KLASSE WÄHREND DER ZAHNENTWICKLUNG (LETZTE HÄLFTE DER SCHWANGERSCHAFT, KINDHEIT UND KINDHEIT BIS ZUM ALTER VON 8 JAHREN) KANN ZU DAUERHAFTEN VERFÄRBUNGEN DER ZÄHNE (GELB-GRAU-BRAUN) FÜHREN.
Diese Nebenwirkung tritt häufiger bei Langzeitanwendung der Arzneimittel auf, wurde jedoch nach wiederholten Kurzzeitzyklen beobachtet. Es wurde auch über Schmelzhypoplasie berichtet. ARZNEIMITTEL MIT TETRACYCLIN SOLLTEN DESHALB WÄHREND DER ZAHNENTWICKLUNG NICHT VERWENDET WERDEN, ES SEI DENN, ANDERE ARZNEIMITTEL SIND WAHRSCHEINLICH NICHT WIRKSAM ODER KONTRAINDIZIERT.
Alle Tetracycline bilden in allen knochenbildenden Geweben einen stabilen Calciumkomplex. Bei jungen Tieren (Ratten und Kaninchen), denen Tetracyclin in Dosen von 25 mg/kg alle sechs Stunden oral verabreicht wurde, wurde eine Abnahme der Fibulawachstumsrate beobachtet. Diese Reaktion erwies sich als reversibel, wenn das Medikament abgesetzt wurde.
Ergebnisse von Tierversuchen weisen darauf hin, dass Tetracycline die Plazenta passieren, in fötalen Geweben gefunden werden und toxische Wirkungen auf den sich entwickelnden Fötus haben können (häufig im Zusammenhang mit einer Verzögerung der Skelettentwicklung). Hinweise auf Embryotoxizität wurden auch bei Tieren festgestellt, die früh in der Trächtigkeit behandelt wurden.
Die antianabole Wirkung von Tetracyclin kann zu einem Anstieg von BUN führen. Während dies bei Patienten mit normaler Nierenfunktion kein Problem darstellt, können höhere Serumspiegel von Tetracyclin bei Patienten mit erheblich eingeschränkter Funktion zu Azotämie, Hyperphosphatämie und Azidose führen. Wenn eine Nierenfunktionsstörung vorliegt, kann sogar die übliche orale oder parenterale Dosis zu einer übermäßigen systemischen Akkumulation des Arzneimittels und einer möglichen Lebertoxizität führen. Unter solchen Bedingungen sind niedrigere Dosen als üblich angezeigt, und wenn die Therapie verlängert wird, können Bestimmungen des Serumspiegels des Arzneimittels ratsam sein.
Lichtempfindlichkeit, die sich in einer übertriebenen Sonnenbrandreaktion äußert, wurde bei einigen Personen beobachtet, die Tetracycline einnahmen. Patienten, die dazu neigen, direktem Sonnenlicht oder ultraviolettem Licht ausgesetzt zu sein, sollten darauf hingewiesen werden, dass diese Reaktion bei Tetracyclin-Medikamenten auftreten kann, und die Behandlung sollte beim ersten Anzeichen eines Hauterythems abgebrochen werden.
HINWEIS: Photosensibilisierungsreaktionen traten am häufigsten bei Demeclocyclin auf, weniger bei Chlortetracyclin und sehr selten bei Oxytetracyclin und Tetracyclin.
VORSICHTSMASSNAHMEN
Allgemein
Es ist unwahrscheinlich, dass die Verschreibung von Sumycin '250' und Sumycin '500' Tabletten (Tetracyclinhydrochlorid-Tabletten) ohne nachgewiesene oder dringend vermutete bakterielle Infektion oder eine prophylaktische Indikation dem Patienten einen Nutzen bringt und das Risiko der Entwicklung einer Arzneimittelresistenz erhöht Bakterien.
Wie bei anderen Antibiotika kann die Anwendung dieses Arzneimittels zu einem übermäßigen Wachstum von nicht empfindlichen Organismen, einschließlich Pilzen, führen. Wenn eine Superinfektion auftritt, sollte das Antibiotikum abgesetzt und eine geeignete Therapie eingeleitet werden. HINWEIS: Eine Superinfektion des Darms durch Staphylokokken kann lebensbedrohlich sein.
Pseudotumor cerebri (benigne intrakranielle Hypertonie) bei Erwachsenen wurde mit der Anwendung von Tetracyclinen in Verbindung gebracht. Die üblichen klinischen Manifestationen sind Kopfschmerzen und verschwommenes Sehen. Hervortretende Fontanellen wurden mit der Anwendung von Tetracyclinen bei Säuglingen in Verbindung gebracht. Während diese beiden Zustände und die damit verbundenen Symptome in der Regel nach Absetzen des Tetracyclins verschwinden, besteht die Möglichkeit dauerhafter Folgeerscheinungen.
Da Überempfindlichkeitsreaktionen eher bei Personen mit Allergien, Asthma, Heuschnupfen oder Urtikaria in der Vorgeschichte auftreten, sollte das Präparat bei solchen Personen mit Vorsicht angewendet werden.
Eine Kreuzsensibilisierung zwischen den verschiedenen Tetracyclinen ist sehr häufig.
Inzision und Drainage oder andere chirurgische Eingriffe sollten, wenn angezeigt, in Verbindung mit einer Antibiotikatherapie durchgeführt werden.
Unter keinen Umständen sollten veraltete Tetracycline verabreicht werden, da der Abbau von Tetracyclinen hochgradig nephrotoxisch ist und gelegentlich ein Fanconi-ähnliches Syndrom hervorgerufen hat.
Labortests
Während der Langzeittherapie sollten regelmäßige Laboruntersuchungen der Funktion des Organsystems, einschließlich der Nieren, der Leber und des hämatopoetischen Systems, durchgeführt werden.
Alle Patienten mit Gonorrhoe sollten sich zum Zeitpunkt der Diagnose einem serologischen Test auf Syphilis unterziehen. Patienten, die mit Tetracyclin behandelt werden, sollten nach 3 Monaten einen serologischen Follow-up-Test auf Syphilis haben.
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Langzeitstudien an Ratten und Mäusen zur Feststellung, ob Tetracyclinhydrochlorid ein kanzerogenes Potenzial hat, waren negativ. Einige verwandte Antibiotika (Oxytetracyclin, Minocyclin) haben Hinweise auf eine onkogene Aktivität bei Ratten gezeigt. In zwei In-vitro-Untersuchungssystemen mit Säugetierzellen (L51784y-Lymphom der Maus und Lungenzellen des chinesischen Hamsters) gab es Hinweise auf Mutagenität bei Tetracyclin-Hydrochlorid-Konzentrationen von 60 bzw. 10 µg/ml.
Tetracyclinhydrochlorid hatte keine Auswirkung auf die Fertilität, wenn es männlichen und weiblichen Ratten mit einer täglichen Aufnahme in Höhe des 25-fachen der menschlichen Dosis über das Futter verabreicht wurde.
Schwangerschaft: Teratogene Wirkungen: Schwangerschaftskategorie D (vgl WARNUNGEN. )
Schwangerschaft: Nicht teratogene Wirkungen: (sehen WARNUNGEN. )
Arbeit und Lieferung
Die Wirkung von Tetracyclinen auf Wehen und Geburt ist nicht bekannt.
Stillende Mutter
Tetracycline sind in der Milch stillender Frauen vorhanden, die ein Medikament dieser Klasse einnehmen. Aufgrund der Möglichkeit schwerwiegender Nebenwirkungen von Tetracyclinen beim gestillten Säugling sollte eine Entscheidung getroffen werden, ob das Stillen oder das Medikament abgesetzt werden soll, wobei die Bedeutung des Medikaments für die Mutter zu berücksichtigen ist (siehe Abschnitt 4.4). WARNUNGEN .)
Pädiatrische Verwendung
Sehen WARNUNGEN und DOSIERUNG UND ANWENDUNG.
ÜBERDOSIS
Im Falle einer Überdosierung symptomatisch behandeln und unterstützende Maßnahmen einleiten.
KONTRAINDIKATIONEN
Dieses Medikament ist kontraindiziert bei Personen, die eine Überempfindlichkeit gegenüber einem der Tetracycline gezeigt haben.
KLINISCHE PHARMAKOLOGIE
Tetracycline werden ausreichend, aber unvollständig aus dem Gastrointestinaltrakt resorbiert. Ungefähr 65 Prozent eines kurzwirksamen Tetracyclins sind an Plasmaproteine gebunden; die Plasmaproteinbindung für mittellang und lang wirkende Analoga ist normalerweise größer.
Die Penetration der Tetracycline in die meisten Körperflüssigkeiten und Gewebe ist ausgezeichnet. Tetracycline werden in unterschiedlichem Ausmaß in Galle, Leber, Lunge, Niere, Prostata, Urin, Liquor cerebrospinalis, Synovialflüssigkeit, Schleimhaut der Kieferhöhle, Gehirn, Sputum und Knochen verteilt. Tetracycline passieren die Plazenta und gelangen in den fötalen Kreislauf und das Fruchtwasser.
Nach einer oralen Einzeldosis werden maximale Plasmakonzentrationen innerhalb von zwei bis vier Stunden erreicht.
Tetracycline werden von der Leber in der Galle konzentriert. Sie werden sowohl im Urin als auch im Kot in hoher Konzentration in biologisch aktiver Form ausgeschieden. Da die renale Clearance von Tetracyclinen durch glomeruläre Filtration erfolgt, wird die Ausscheidung signifikant durch den Zustand der Nierenfunktion beeinflusst. (Sehen WARNUNGEN. )
Mikrobiologie
Die Tetracycline sind in erster Linie bakteriostatisch und sollen ihre antimikrobielle Wirkung über die Hemmung der Proteinsynthese ausüben. Die Tetracycline haben ein ähnliches antimikrobielles Wirkungsspektrum gegen ein breites Spektrum grampositiver und gramnegativer Organismen. Kreuzresistenzen dieser Organismen gegenüber Tetracyclinen sind häufig. Darüber hinaus können gramnegative Bazillen, die Tetracyclin-resistent gemacht wurden, auch eine Kreuzresistenz gegen Chloramphenicol aufweisen.
GRAM-NEGATIVE BAKTERIEN:
Bartonella bacilliformis Brucella-Spezies Calymmatobacterium granulomatis Campylobacter fetus Francisella tularensis Haemophilus ducreyi Haemophilus influenzae Listeria monocytogenes Neisseria gonorrhoeae Vibrio cholerae Yersinia pestis
Da sich viele Stämme der folgenden Gruppen gramnegativer Mikroorganismen als resistent gegenüber Tetrazyklinen erwiesen haben, sind Kultur- und Empfindlichkeitstests besonders zu empfehlen:
Acinetobacter-Spezies Bacteroides-Spezies Enterobacter aerogenes Escherichia coli Klebsiella-Spezies Shigella-Spezies
GRAM-POSITIVE BAKTERIEN:
Enterococcus-Gruppe [Enterococcus faecalis, (früher Streptococcus faecalis) und Enterococcus faecium (früher Streptococcus faecium)
Streptococci viridans Gruppe Streptococcus pneumoniae Streptococcus pyogenes
Da sich viele Stämme dieser grampositiven Mikroorganismen als resistent gegen Tetracyclin erwiesen haben, werden Kultur- und Empfindlichkeitstests empfohlen. Bis zu 44 Prozent der Stämme von Streptococcus pyogenes und 74 Prozent von Enterococcus faecalis (ehemals Streptococcus faecalis) haben sich als resistent gegen Tetracyclin-Medikamente erwiesen. Daher sollten Tetracycline nicht zur Behandlung von Streptokokken-Erkrankungen verwendet werden, es sei denn, der Organismus ist bekanntermaßen empfindlich.
ANDERE MIKROORGANISMEN:
Actinomyces-Arten Bacillus anthracis Balantidium coli Borrelia recurrentis Chlamydia psittaci Chlamydia trachomatis Clostridium-Arten Entamoeba-Arten Fusobacterium fusiforme Mycoplasma pneumoniae Rickettsiae Propionibacterium acnes Treponema pallidum Treponema pertenue Ureaplasma urealyticum
Empfindlichkeitsprüfung
Diffusionstechniken
Quantitative Methoden, die die Messung von Zonendurchmessern erfordern, geben die genaueste Schätzung der Empfindlichkeit von Bakterien gegenüber antimikrobiellen Mitteln.
Ein solches Standardverfahren1, das zur Verwendung mit Blättchen empfohlen wurde, um die Empfindlichkeit von Mikroorganismen gegenüber Tetracyclin zu testen, verwendet das 30-µg-Tetracyclin-Blättchen. Zur Interpretation gehört die Korrelation der im Scheibentest ermittelten Zonendurchmesser mit der minimalen Hemmkonzentration (MHK) für Tetracyclin.
Berichte aus dem Labor, die Ergebnisse des standardmäßigen Einzelscheiben-Empfindlichkeitstests mit einer 30-µg-Tetracyclin-Scheibe enthalten, sollten gemäß den folgenden Kriterien interpretiert werden:
Ein Bericht von „anfällig“ weist darauf hin, dass der Erreger wahrscheinlich durch allgemein erreichbare Blutspiegel gehemmt wird. Ein Bericht von „Intermediate“ deutet darauf hin, dass der Organismus anfällig wäre, wenn eine hohe Dosierung verwendet wird oder wenn die Infektion auf Gewebe oder Flüssigkeiten beschränkt ist, in denen hohe antibiotische (oder antimikrobielle) Konzentrationen erreicht werden. Ein Bericht von „Resistenz“ weist darauf hin, dass erreichbare Konzentrationen wahrscheinlich nicht hemmend wirken und eine andere Therapie gewählt werden sollte.
Standardisierte Verfahren erfordern die Verwendung von Laborkontrollorganismen. Die 30-µg-Tetracyclin-Scheibe sollte die folgenden Zonendurchmesser ergeben:
Verdünnungstechniken
Verwenden Sie eine standardisierte Verdünnungsmethode2 (Brühe, Agar, Mikroverdünnung) oder eine gleichwertige Methode mit Tetracyclin-Pulver. Die erhaltenen MHK-Werte sollten nach folgenden Kriterien interpretiert werden:
Wie bei Standard-Diffusionstechniken erfordern Verdünnungsmethoden die Verwendung von Laborkontrollorganismen. Standard-Tetracyclin-Pulver sollte folgende MHK-Werte aufweisen:
Tierpharmakologie und Tiertoxikologie
Hyperpigmentierung der Schilddrüse wurde durch Mitglieder der Tetracyclin-Klasse bei den folgenden Spezies hervorgerufen: bei Ratten durch Oxytetracyclin, Doxycyclin, Tetracyclin PO4 und Methacyclin; bei Minischweinen durch Doxycyclin, Minocyclin, Tetracyclin PO4 und Methacyclin; bei Hunden durch Doxycyclin und Minocyclin; bei Affen durch Minocyclin.
Minocyclin, Tetracyclin PO4, Methacyclin, Doxycyclin, Tetracyclin-Base, Oxytetracyclin-HCl und Tetracyclin-HCl waren goitrogen bei Ratten, denen eine jodarme Diät verabreicht wurde. Diese goitrogene Wirkung wurde von einer hohen radioaktiven Jodaufnahme begleitet. Die Verabreichung von Minocyclin erzeugte auch einen großen Kropf mit hoher Radiojodaufnahme bei Ratten, die mit einer relativ jodreichen Diät gefüttert wurden.
Die Behandlung verschiedener Tierspezies mit dieser Arzneimittelklasse führte auch bei folgenden Tieren zur Induktion von Schilddrüsenhyperplasien: bei Ratten und Hunden (Minocyclin), bei Hühnern (Chlortetracyclin) und bei Ratten und Mäusen (Oxytetracyclin). Bei mit Oxytetracyclin behandelten Ziegen und Ratten wurde eine Nebennierenhyperplasie beobachtet.
HINWEIS
1. National Committee for Clinical Laboratory Standards, Performance Standards for Antimicrobial Disk Susceptibility Tests – Fourth Edition. Genehmigtes Standard-NCCLS-Dokument M2-A4, Vol. 10, Nr. 7 NCCLS, Villanova, PA, April 1990.
2. National Committee for Clinical Laboratory Standards, Methods for Dilution Antimicrobial susceptibility tests for Bacteria that grow aerobically-Second Edition. Genehmigtes Standard-NCCLS-Dokument M7-A2, Vol. 10, Nr. 8 NCCLS, Villanova, PA, April 1990.
INFORMATIONEN ZUM PATIENTEN
Die Patienten sollten darauf hingewiesen werden, dass antibakterielle Arzneimittel einschließlich Sumycin '250' und Sumycin '500' Tabletten (Tetracyclinhydrochlorid-Tabletten) nur zur Behandlung bakterieller Infektionen verwendet werden sollten. Sie behandeln keine Virusinfektionen (z. B. Erkältung). Wenn Sumycin '250' und Sumycin '500' Tabletten (Tetracyclin-Hydrochlorid-Tabletten) zur Behandlung einer bakteriellen Infektion verschrieben werden, sollten die Patienten darüber informiert werden, dass sie sich zwar zu Beginn der Therapie besser fühlen, das Medikament jedoch genau so eingenommen werden sollte gerichtet. Das Auslassen von Dosen oder das Nichtbeenden des vollständigen Behandlungszyklus kann (1) die Wirksamkeit der sofortigen Behandlung verringern und (2) die Wahrscheinlichkeit erhöhen, dass Bakterien Resistenzen entwickeln und mit Sumycin '250' und Sumycin '500' Tabletten nicht behandelbar sind ( Tetracyclin-Hydrochlorid-Tabletten) oder andere antibakterielle Arzneimittel in der Zukunft.
Was ist Suprax 200 mg und wie wird es angewendet?
Suprax (Cefixim) zur oralen Suspension ist ein Cephalosporin-Antibiotikum, das zur Behandlung vieler verschiedener Arten von Infektionen verwendet wird, die durch Bakterien verursacht werden.
Welche Nebenwirkungen hat Suprax?
Häufige Nebenwirkungen von Suprax 100 mg sind:
Informieren Sie Ihren Arzt, wenn bei Ihnen seltene, aber sehr schwerwiegende Nebenwirkungen von Suprax 200 mg auftreten, einschließlich:
BEZEICHNUNG
Cefixim ist ein halbsynthetisches Cephalosporin-Antibiotikum zur oralen Verabreichung. Chemisch ist es (6R,7R)-7-[2-(2-Amino-4-thiazolyl)glyoxylamido]-8-oxo-3-vinyl-5-thia-1-azabicyclo[4.2.0]oct-2 -en-2-carbonsäure, 72-(Z)-[O-(Carboxymethyl)oxim]trihydrat.
Molekulargewicht = 507,50 als Trihydrat. Die chemische Formel ist C16H15N5O7S2.3H2O
Die Strukturformel für Cefixim lautet:
INDIKATIONEN
Unkomplizierte Harnwegsinfektionen
SUPRAX ist indiziert zur Behandlung von Erwachsenen und pädiatrischen Patienten ab einem Alter von sechs Monaten mit unkomplizierten Harnwegsinfektionen, die durch empfindliche Isolate von Escherichia coli und Proteus mirabilis verursacht werden.
Mittelohrentzündung
SUPRAX ist indiziert zur Behandlung von Erwachsenen und pädiatrischen Patienten ab einem Alter von sechs Monaten mit Mittelohrentzündung, die durch empfindliche Isolate von Haemophilus influenzae, Moraxella catarrhalis und Streptococcus pyogenes verursacht wird. (Die Wirksamkeit für Streptococcus pyogenes in diesem Organsystem wurde bei weniger als 10 Infektionen untersucht.)
Notiz
Bei Patienten mit Mittelohrentzündung, die durch Streptococcus pneumoniae verursacht wurde, war das Gesamtansprechen auf Cefixim ungefähr 10 % geringer als auf das Vergleichspräparat [siehe Klinische Studien ].
Pharyngitis und Mandelentzündung
SUPRAX ist indiziert zur Behandlung von Erwachsenen und pädiatrischen Patienten ab einem Alter von sechs Monatenmit Pharyngitis und Tonsillitis, die durch empfindliche Isolate von Streptococcus pyogenes verursacht werden. (Anmerkung: Penicillin ist das übliche Mittel der Wahl bei der Behandlung von Infektionen mit Streptococcus pyogenes. SUPRAX ist im Allgemeinen wirksam bei der Eradikation von Streptococcus pyogenes aus dem Nasopharynx; Daten, die die Wirksamkeit von SUPRAX 100 mg bei der anschließenden Prävention von rheumatischem Fieber belegen, sind dies jedoch nicht verfügbar.)
Akute Exazerbationen einer chronischen Bronchitis
SUPRAX 200 mg ist indiziert zur Behandlung von Erwachsenen und pädiatrischen Patienten ab einem Alter von sechs Monaten mit akuten Exazerbationen einer chronischen Bronchitis, die durch empfindliche Isolate von Streptococcus pneumoniae und Haemophilus influenzae verursacht werden.
Unkomplizierte Gonorrhoe (zervikal/urethral)
SUPRAX 100 mg ist indiziert zur Behandlung von Erwachsenen und pädiatrischen Patienten ab einem Alter von sechs Monaten mit unkomplizierter Gonorrhö (zervikal/urethral), die durch empfindliche Isolate von Neisseria gonorrhoeae (Penicillinase- und nicht-Penicillinase-produzierende Isolate) verursacht wird.
Verwendungszweck
Um die Entwicklung arzneimittelresistenter Bakterien zu verringern und die Wirksamkeit von SUPRAX 100 mg und anderen antibakteriellen Arzneimitteln aufrechtzuerhalten, sollte SUPRAX 100 mg nur zur Behandlung von Infektionen angewendet werden, die nachweislich oder mit starkem Verdacht auf empfindliche Bakterien zurückzuführen sind. Wenn Kultur- und Empfindlichkeitsinformationen verfügbar sind, sollten diese bei der Auswahl oder Änderung der antimikrobiellen Therapie berücksichtigt werden. In Ermangelung solcher Daten können lokale Epidemiologie und Anfälligkeitsmuster zur empirischen Auswahl der Therapie beitragen.
DOSIERUNG UND ANWENDUNG
Erwachsene
Die empfohlene Dosis von Cefixim beträgt 400 mg täglich. Dies kann als 400-mg-Tablette oder -Kapsel täglich gegeben werden, oder die 400-mg-Tablette kann geteilt und als halbe Tablette alle 12 Stunden gegeben werden. Zur Behandlung unkomplizierter zervikaler/urethraler Gonokokkeninfektionen wird eine orale Einzeldosis von 400 mg empfohlen. Die Kapsel und Tablette kann unabhängig von Nahrung verabreicht werden.
Bei der Behandlung von Infektionen durch Streptococcus pyogenes sollte eine therapeutische Dosis von Cefixim für mindestens 10 Tage verabreicht werden.
Pädiatrische Patienten (6 Monate oder älter)
Die empfohlene Dosis beträgt 8 mg/kg/Tag der Suspension. Dies kann als einzelne Tagesdosis verabreicht werden oder kann in zwei getrennten Dosen als 4 mg/kg alle 12 Stunden verabreicht werden.
Notiz
Für jeden pädiatrischen Gewichtsbereich wurde eine empfohlene Dosis festgelegt. Siehe Tabelle 1. Stellen Sie sicher, dass alle Bestellungen, die eine Dosis in Millilitern angeben, eine Konzentration enthalten, da SUPRAX zur Herstellung einer Suspension zum Einnehmen in drei verschiedenen Konzentrationen erhältlich ist (100 mg/5 ml, 200 mg/5 ml und 500 mg/5 ml).
Kinder, die mehr als 45 kg wiegen oder älter als 12 Jahre sind, sollten mit der empfohlenen Erwachsenendosis behandelt werden. SUPRAX (Cefixim) Kautabletten müssen vor dem Schlucken gekaut oder zerkleinert werden.
Otitis media sollte mit den Kautabletten oder der Suspension behandelt werden. Klinische Studien zu Mittelohrentzündung wurden mit den Kautabletten oder der Suspension durchgeführt, und die Kautabletten oder die Suspension führen zu höheren maximalen Blutspiegeln als die Tablette, wenn sie in derselben Dosis verabreicht werden.
Daher sollte die Tablette oder Kapsel die Kautabletten oder Suspension bei der Behandlung von Mittelohrentzündung nicht ersetzen [siehe KLINISCHE PHARMAKOLOGIE ].
Bei der Behandlung von Infektionen durch Streptococcus pyogenes sollte eine therapeutische Dosis von Cefixim für mindestens 10 Tage verabreicht werden.
Nierenfunktionsstörung
SUPRAX 200 mg kann bei eingeschränkter Nierenfunktion verabreicht werden. Bei Patienten mit einer Kreatinin-Clearance von 60 ml/min oder mehr können die normale Dosis und das normale Behandlungsschema angewendet werden. Siehe Tabelle 2 für Dosisanpassungen bei Erwachsenen mit eingeschränkter Nierenfunktion. Weder die Hämodialyse noch die Peritonealdialyse entfernen signifikante Arzneimittelmengen aus dem Körper.
Rekonstitutionsanweisungen für die orale Suspension
Nach der Rekonstitution kann die Suspension 14 Tage lang entweder bei Raumtemperatur oder im Kühlschrank ohne signifikanten Wirkungsverlust aufbewahrt werden. Dicht verschlossen halten. Vor Gebrauch gut schütteln. Verwerfen Sie nicht verwendete Portionen nach 14 Tagen.
WIE GELIEFERT
Darreichungsformen und Stärken
SUPRAX ist zur oralen Verabreichung in den folgenden Darreichungsformen und Stärken erhältlich:
Lagerung und Handhabung
SUPRAX® ist zur oralen Verabreichung in den folgenden Dosierungsformen, Stärken und Packungen erhältlich, die in der folgenden Tabelle aufgeführt sind:
Hergestellt von: Lupine Limited Mandideep 462 046 Indien. Überarbeitet: März 2018.
NEBENWIRKUNGEN
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Die am häufigsten beobachteten Nebenwirkungen in US-amerikanischen Studien zur Tablettenformulierung waren gastrointestinale Ereignisse, die bei 30 % der erwachsenen Patienten entweder bei zweimal täglicher oder einmal täglicher Einnahme berichtet wurden. Fünf Prozent (5 %) der Patienten in den klinischen Studien in den USA brachen die Therapie wegen arzneimittelbedingter Nebenwirkungen ab. Einzelne Nebenwirkungen waren Durchfall 16 %, weicher oder häufiger Stuhlgang 6 %, Bauchschmerzen 3 %, Übelkeit 7 %, Dyspepsie 3 % und Flatulenz 4 %. Die Inzidenz von gastrointestinalen Nebenwirkungen, einschließlich Durchfall und weichem Stuhl, bei pädiatrischen Patienten, die die Suspension erhielten, war vergleichbar mit der Inzidenz, die bei erwachsenen Patienten beobachtet wurde, die Tabletten erhielten.
Post-Marketing-Erfahrung
Die folgenden Nebenwirkungen wurden nach der Anwendung von Cefixim nach der Zulassung berichtet. Die Inzidenzraten betrugen weniger als 1 von 50 (weniger als 2 %).
Magen-Darm
In klinischen Studien wurden mehrere Fälle von dokumentierter pseudomembranöser Kolitis identifiziert. Das Auftreten pseudomembranöser Colitis-Symptome kann während oder nach der Therapie auftreten.
Überempfindlichkeitsreaktionen
Anaphylaktische/anaphylaktoide Reaktionen (einschließlich Schock und Todesfälle), Hautausschläge, Urtikaria, Arzneimittelfieber, Pruritus, Angioödem und Gesichtsödem. Erythema multiforme, Stevens-Johnson-Syndrom und Serumkrankheit-ähnliche Reaktionen wurden berichtet.
Leber
Rachenartige Erhöhungen von SGPT, SGOT, alkalischer Phosphatase, Hepatitis, Gelbsucht.
Nieren
Vorübergehende Erhöhungen von BUN oder Kreatinin, akutes Nierenversagen.
Zentrales Nervensystem
Kopfschmerzen, Schwindel, Krampfanfälle.
Hämisches und lymphatisches System
Vorübergehende Thrombozytopenie, Leukopenie, Neutropenie, Verlängerung der Prothrombinzeit, erhöhtes LDH, Panzytopenie, Agranulozytose und Eosinophilie.
Abnormale Labortests
Hyperbilirubinämie.
Andere Nebenwirkungen
Genitaler Pruritus, Vaginitis, Candidiasis, toxische epidermale Nekrolyse.
Gemeldete Nebenwirkungen für Arzneimittel der Cephalosporin-Klasse
Allergische Reaktionen, Superinfektion, Nierenfunktionsstörung, toxische Nephropathie, Leberfunktionsstörung einschließlich Cholestase, aplastische Anämie, hämolytische Anämie, Blutungen und Colitis.
Mehrere Cephalosporine wurden mit der Auslösung von Krampfanfällen in Verbindung gebracht, insbesondere bei Patienten mit eingeschränkter Nierenfunktion, wenn die Dosierung nicht reduziert wurde [siehe DOSIERUNG UND ANWENDUNG sehen ÜBERDOSIS ]. Wenn im Zusammenhang mit der medikamentösen Therapie Krampfanfälle auftreten, sollte das Medikament abgesetzt werden. Bei klinischer Indikation kann eine antikonvulsive Therapie gegeben werden.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Carbamazepin
Nach Markteinführung wurde über erhöhte Carbamazepin-Spiegel bei gleichzeitiger Anwendung von Cefixim berichtet. Die Überwachung von Arzneimitteln kann hilfreich sein, um Veränderungen der Carbamazepin-Plasmakonzentrationen zu erkennen.
Warfarin und Antikoagulantien
Bei gleichzeitiger Gabe von Cefixim wurde über eine verlängerte Prothrombinzeit mit oder ohne klinische Blutung berichtet.
Arzneimittel-/Labortest-Wechselwirkungen
Eine falsch-positive Reaktion auf Ketone im Urin kann bei Tests auftreten, die Nitroprussid verwenden, aber nicht bei Tests, die Nitroferricyanid verwenden.
Die Verabreichung von Cefixim kann mit Clinitest®, Benedict-Lösung oder Fehling-Lösung zu einer falsch-positiven Reaktion auf Glukose im Urin führen. Es wird empfohlen, Glukosetests auf Basis enzymatischer Glukoseoxidase-Reaktionen (wie Clinistix® oder TesTape®) zu verwenden. Während der Behandlung mit anderen Cephalosporinen wurde über einen falsch positiven direkten Coombs-Test berichtet; Daher sollte berücksichtigt werden, dass ein positiver Coombs-Test auf das Medikament zurückzuführen sein kann.
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Überempfindlichkeitsreaktionen
Anaphylaktische/anaphylaktoide Reaktionen (einschließlich Schock und Todesfälle) wurden bei der Anwendung von Cefixim berichtet.
Bevor eine Therapie mit SUPRAX eingeleitet wird, sollte sorgfältig abgeklärt werden, ob beim Patienten frühere Überempfindlichkeitsreaktionen auf Cephalosporine, Penicilline oder andere Arzneimittel aufgetreten sind. Wenn dieses Produkt Penicillin-empfindlichen Patienten verabreicht werden soll, ist Vorsicht geboten, da eine Kreuzallergie zwischen Beta-Lactam-Antibiotika eindeutig dokumentiert wurde und bei bis zu 10 % der Patienten mit Penicillin-Allergie in der Vorgeschichte auftreten kann. Wenn eine allergische Reaktion auf SUPRAX 100 mg auftritt, setzen Sie das Medikament ab.
Clostridium difficile-assoziierter Durchfall
Clostridium difficile-assoziierte Diarrhoe (CDAD) wurde bei der Anwendung von fast allen antibakteriellen Mitteln, einschließlich SUPRAX 200 mg, berichtet und kann in ihrer Schwere von leichter Diarrhoe bis hin zu tödlicher Colitis reichen. Die Behandlung mit antibakteriellen Mitteln verändert die normale Flora des Dickdarms, was zu einem übermäßigen Wachstum von C. difficile führt.
C. difficile produziert die Toxine A und B, die zur Entstehung von CDAD beitragen. Hypertoxin produzierende Isolate von C. difficile verursachen eine erhöhte Morbidität und Mortalität, da diese Infektionen gegenüber einer antimikrobiellen Therapie refraktär sein können und eine Kolektomie erfordern können. CDAD muss bei allen Patienten in Betracht gezogen werden, die nach der Anwendung antibakterieller Medikamente an Durchfall leiden. Eine sorgfältige Anamnese ist erforderlich, da berichtet wurde, dass CDAD mehr als zwei Monate nach der Verabreichung von Antibiotika auftritt.
Wenn CDAD vermutet oder bestätigt wird, muss die laufende Anwendung antibakterieller Arzneimittel, die nicht gegen C. difficile gerichtet sind, möglicherweise eingestellt werden. Je nach klinischer Indikation sollte ein angemessenes Flüssigkeits- und Elektrolytmanagement, eine Proteinergänzung, eine antibakterielle medikamentöse Behandlung von C. difficile und eine chirurgische Untersuchung eingeleitet werden.
Dosisanpassung bei eingeschränkter Nierenfunktion
Die Dosis von SUPRAX 100 mg sollte bei Patienten mit eingeschränkter Nierenfunktion sowie bei Patienten, die sich einer kontinuierlichen ambulanten Peritonealdialyse (CAPD) und Hämodialyse (HD) unterziehen, angepasst werden. Dialysepatienten sollten sorgfältig überwacht werden [siehe DOSIERUNG UND ANWENDUNG ].
Gerinnungseffekte
Cephalosporine, einschließlich SUPRAX 200 mg, können mit einem Abfall der Prothrombinaktivität in Verbindung gebracht werden. Zu den Risikogruppen gehören Patienten mit eingeschränkter Nieren- oder Leberfunktion oder schlechtem Ernährungszustand sowie Patienten, die eine langwierige antimikrobielle Therapie erhalten, und Patienten, die zuvor mit einer Antikoagulanzientherapie stabilisiert wurden. Bei Risikopatienten sollte die Prothrombinzeit überwacht und exogenes Vitamin K wie angezeigt verabreicht werden.
Entwicklung arzneimittelresistenter Bakterien
Die Verschreibung von SUPRAX (Cefixim) ohne nachgewiesene oder dringend vermutete bakterielle Infektion bringt dem Patienten wahrscheinlich keinen Nutzen und erhöht das Risiko der Entwicklung arzneimittelresistenter Bakterien.
Risiko bei Patienten mit Phenylketonurie
Phenylalanin kann für Patienten mit Phenylketonurie (PKU) schädlich sein. SUPRAX 200 mg Kautabletten enthalten Aspartam, eine Quelle für Phenylalanin. Jede 100-mg-, 150-mg- und 200-mg-Stärke enthält 3,3 mg, 5 mg bzw. 6,7 mg Phenylalanin. Bevor Sie einem Patienten mit PKU SUPRAX Kautabletten verschreiben, sollten Sie die kombinierte tägliche Menge an Phenylalanin aus allen Quellen berücksichtigen, einschließlich SUPRAX 200 mg Kautabletten.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Lebenszeitstudien an Tieren zur Bewertung des kanzerogenen Potenzials wurden nicht durchgeführt. Cefixim verursachte in vitro keine Punktmutationen in Bakterien oder Säugerzellen, DNA-Schäden oder Chromosomenschäden und zeigte in vivo im Maus-Mikrokerntest kein klastogenes Potenzial. Bei Ratten wurden Fertilität und Reproduktionsleistung durch Cefixim in Dosen bis zum 25-Fachen der therapeutischen Dosis für Erwachsene nicht beeinträchtigt.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschaftskategorie B
Reproduktionsstudien wurden an Mäusen und Ratten mit bis zum 40-fachen der menschlichen Dosis durchgeführt und ergaben keine Hinweise auf eine Schädigung des Fötus durch Cefixim. Es liegen keine adäquaten und gut kontrollierten Studien bei Schwangeren vor. Da Reproduktionsstudien an Tieren die menschliche Reaktion nicht immer vorhersagen, sollte dieses Medikament während der Schwangerschaft nur angewendet werden, wenn dies eindeutig erforderlich ist.
Arbeit und Lieferung
Cefixim wurde nicht für die Anwendung während der Wehen und der Geburt untersucht. Eine Behandlung sollte nur erfolgen, wenn dies eindeutig erforderlich ist.
Stillende Mutter
Es ist nicht bekannt, ob Cefixim in die Muttermilch übergeht. Es sollte erwogen werden, das Stillen während der Behandlung mit diesem Arzneimittel vorübergehend zu unterbrechen.
Pädiatrische Verwendung
Sicherheit und Wirksamkeit von Cefixim bei Kindern unter sechs Monaten sind nicht erwiesen. Die Inzidenz von gastrointestinalen Nebenwirkungen, einschließlich Durchfall und weichem Stuhl, bei pädiatrischen Patienten, die die Suspension erhielten, war vergleichbar mit der Inzidenz, die bei erwachsenen Patienten beobachtet wurde, die Tabletten erhielten.
Geriatrische Verwendung
Klinische Studien umfassten keine ausreichende Anzahl von Probanden im Alter von 65 Jahren und älter, um festzustellen, ob sie anders reagieren als jüngere Probanden. Andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt. In einer pharmakokinetischen Studie bei älteren Menschen wurden Unterschiede in den pharmakokinetischen Parametern festgestellt [siehe KLINISCHE PHARMAKOLOGIE ]. Diese Unterschiede waren gering und weisen nicht auf die Notwendigkeit einer Dosisanpassung des Arzneimittels bei älteren Patienten hin.
Nierenfunktionsstörung
Die Dosis von Cefixim sollte bei Patienten mit eingeschränkter Nierenfunktion sowie bei Patienten, die sich einer kontinuierlichen ambulanten Peritonealdialyse (CAPD) und Hämodialyse (HD) unterziehen, angepasst werden. Dialysepatienten sollten sorgfältig überwacht werden [siehe DOSIERUNG UND ANWENDUNG ].
ÜBERDOSIS
Eine Magenspülung kann angezeigt sein; andernfalls existiert kein spezifisches Antidot. Cefixim wird durch Hämodialyse oder Peritonealdialyse nicht in nennenswerten Mengen aus dem Kreislauf entfernt. Die Nebenwirkungen bei einer kleinen Anzahl gesunder erwachsener Probanden, die Einzeldosen von bis zu 2 g Cefixim erhielten, unterschieden sich nicht von dem Profil, das bei Patienten beobachtet wurde, die mit den empfohlenen Dosen behandelt wurden.
KONTRAINDIKATIONEN
Suprax (Cefixim) ist bei Patienten mit bekannter Allergie gegen Cefixim oder andere Cephalosporine kontraindiziert.
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Cefixim ist ein halbsynthetisches Cephalosporin-Antibiotikum [vgl Mikrobiologie ].
Pharmakokinetik
SUPRAX 200 mg Kautabletten sind bioäquivalent zu einer Suspension zum Einnehmen.
SUPRAX 200 mg Tabletten und Suspension werden bei oraler Verabreichung zu etwa 40 % bis 50 % resorbiert, unabhängig davon, ob sie mit oder ohne Nahrung verabreicht werden; jedoch verlängert sich die Zeit bis zur maximalen Resorption um etwa 0,8 Stunden, wenn es zusammen mit Nahrung verabreicht wird. Eine einzelne 200-mg-Tablette Cefixim erzeugt eine durchschnittliche maximale Serumkonzentration von etwa 2 µg/ml (Bereich 1 bis 4 µg/ml); eine einzelne 400-mg-Tablette erzeugt eine durchschnittliche Spitzenkonzentration von etwa 3,7 µg/ml (Bereich 1,3 bis 7,7 µg/ml). Die Suspension zum Einnehmen erzeugt durchschnittliche Spitzenkonzentrationen, die etwa 25 % bis 50 % höher sind als die der Tabletten, wenn sie an normalen erwachsenen Probanden getestet wird. 200-mg-Dosen der Suspension zum Einnehmen erzeugen durchschnittliche Spitzenkonzentrationen von 3 µg/ml (Bereich 1 bis 4,5 µg/ml) bzw. 4,6 µg/ml (Bereich 1,9 bis 7,7 µg/ml), wenn sie an normalen erwachsenen Probanden getestet wurden . Die Fläche unter der Zeit-Konzentrations-Kurve (AUC) ist bei der Suspension zum Einnehmen um etwa 10 % bis 25 % größer als bei der Tablette nach Dosierungen von 100 bis 400 mg, wenn an normalen erwachsenen Probanden getestet wurde. Diese erhöhte Resorption sollte berücksichtigt werden, wenn die Tablette durch die Suspension zum Einnehmen ersetzt werden soll. Aufgrund der fehlenden Bioäquivalenz sollten Tabletten zur oralen Suspension bei der Behandlung von Mittelohrentzündung nicht ersetzt werden [siehe DOSIERUNG UND ANWENDUNG ]. Bei Kindern wurden keine Crossover-Studien von Tabletten versus Suspensionen durchgeführt.
Die 400-mg-Kapsel ist unter Nüchternbedingungen mit der 400-mg-Tablette bioäquivalent. Nahrung reduziert jedoch die Resorption nach Verabreichung der Kapsel um etwa 15 %, basierend auf AUC und 25 %, basierend auf Cmax.
Maximale Serumkonzentrationen treten zwischen 2 und 6 Stunden nach oraler Verabreichung einer einzelnen 200-mg-Tablette, einer einzelnen 400-mg-Tablette oder einer 400-mg-Cefixim-Suspension auf. Maximale Serumkonzentrationen treten zwischen 2 und 5 Stunden nach einer einmaligen Verabreichung von 200 mg Suspension auf. Maximale Serumkonzentrationen treten zwischen 3 und 8 Stunden nach der oralen Verabreichung einer einzelnen 400-mg-Kapsel auf.
Verteilung
Die Serumproteinbindung ist mit einem gebundenen Anteil von etwa 65 % konzentrationsunabhängig. In einer Mehrfachdosisstudie, die mit einer Forschungsformulierung durchgeführt wurde, die weniger bioverfügbar ist als die Tablette oder Suspension, kam es nach 14-tägiger Verabreichung zu einer geringen Akkumulation des Arzneimittels im Serum oder Urin. Ausreichende Daten zu Cefixim-Spiegeln im Liquor sind nicht verfügbar.
Stoffwechsel und Ausscheidung
Es gibt keine Hinweise auf eine Metabolisierung von Cefixim in vivo. Ungefähr 50 % der resorbierten Dosis werden innerhalb von 24 Stunden unverändert mit dem Urin ausgeschieden. In Tierversuchen wurde festgestellt, dass Cefixim in mehr als 10 % der verabreichten Dosis auch in die Galle ausgeschieden wird. Die Serumhalbwertszeit von Cefixim bei gesunden Probanden ist unabhängig von der Darreichungsform und beträgt im Durchschnitt 3 bis 4 Stunden, kann aber bei einigen gesunden Probanden bis zu 9 Stunden betragen.
Besondere Populationen
Geriatrie
Die durchschnittlichen AUCs im Steady State bei älteren Patienten sind etwa 40 % höher als die durchschnittlichen AUCs anderer gesunder Erwachsener. Unterschiede in den pharmakokinetischen Parametern zwischen 12 jungen und 12 älteren Probanden, die 400 mg Cefixim einmal täglich über 5 Tage erhielten, werden wie folgt zusammengefasst:
Diese Erhöhungen waren jedoch klinisch nicht signifikant [vgl DOSIERUNG UND ANWENDUNG ].
Nierenfunktionsstörung
Bei Patienten mit mäßig eingeschränkter Nierenfunktion (20 bis 40 ml/min Kreatinin-Clearance) verlängert sich die durchschnittliche Serumhalbwertszeit von Cefixim auf 6,4 Stunden. Bei schwerer Nierenfunktionsstörung (5 bis 20 ml/min Kreatinin-Clearance) erhöhte sich die Halbwertszeit auf durchschnittlich 11,5 Stunden. Das Arzneimittel wird durch Hämodialyse oder Peritonealdialyse nicht signifikant aus dem Blut entfernt. Eine Studie zeigte jedoch, dass Patienten, die sich einer Hämodialyse unterziehen, bei Dosen von 400 mg ähnliche Blutprofile aufweisen wie Patienten mit einer Kreatinin-Clearance von 21 bis 60 ml/min.
Mikrobiologie
Wirkmechanismus
Wie bei anderen Cephalosporinen resultiert die bakterizide Wirkung von Cefixim aus der Hemmung der Zellwandsynthese. Cefixim ist in Gegenwart bestimmter Beta-Lactamase-Enzyme stabil. Infolgedessen können bestimmte Organismen, die aufgrund des Vorhandenseins von Betalactamasen gegen Penicilline und einige Cephalosporine resistent sind, gegenüber Cefixim empfindlich sein.
Widerstand
Resistenz gegen Cefixim bei Isolaten von Haemophilus influenzae und Neisseria gonorrhoeae ist meistens mit Veränderungen der Penicillin-bindenden Proteine (PBPs) verbunden. Cefixim hat möglicherweise eine begrenzte Aktivität gegen Enterobacteriaceae, die Extended-Spectrum-Beta-Lactamasen (ESBLs) produzieren. Pseudomonas-Arten, Enterococcus-Arten, Streptokokkenstämme der Gruppe D, Listeria monocytogenes, die meisten Staphylokokkenstämme (einschließlich Methicillin-resistente Stämme), die meisten Enterobacter-Artenstämme, die meisten Bacteroides-fragilis-Stämme und die meisten Clostridium-Artenstämme sind resistent gegen Cefixim.
Antimikrobielle Aktivität
Es wurde gezeigt, dass Cefixim sowohl in vitro als auch bei klinischen Infektionen gegen die meisten Isolate der folgenden Mikroorganismen aktiv ist [vgl INDIKATIONEN ].
Grampositive Bakterien
Streptococcus pneumoniae Streptococcus pyogenes
Gramnegative Bakterien
Escherichia coli Haemophilus influenzae Moraxella catarrhalis Neisseria gonorrhoeae Proteus mirabilis
Die folgenden In-vitro-Daten sind verfügbar, ihre klinische Bedeutung ist jedoch nicht bekannt. Mindestens 90 Prozent der folgenden Bakterien weisen eine minimale In-vitro-Hemmkonzentration (MHK) auf, die kleiner oder gleich der Empfindlichkeitsgrenze für Cefixim gegenüber Isolaten ähnlicher Gattungen oder Organismengruppen ist. Die Wirksamkeit von Cefixim bei der Behandlung klinischer Infektionen, die durch diese Bakterien verursacht werden, wurde jedoch nicht in angemessenen und gut kontrollierten klinischen Studien nachgewiesen.
Grampositive Bakterien
Streptococcus agalactiae
Gramnegative Bakterien
Citrobacter amalonaticus Citrobacter diversus Haemophilus parainfluenzae Klebsiella oxytoca Klebsiella pneumoniae Pasteurella multocida Proteus vulgaris Providencia-Spezies Salmonella-Spezies Serratia marcescens Shigella-Spezies
Empfindlichkeitsprüfung
Spezifische Informationen zu den Interpretationskriterien von Empfindlichkeitstests und zugehörigen Testmethoden und Qualitätskontrollstandards, die von der FDA für dieses Medikament anerkannt sind, finden Sie unter: https://www.fda.gov/STIC.
Klinische Studien
Vergleichende klinische Studien zu Mittelohrentzündung wurden an fast 400 Kindern im Alter von 6 Monaten bis 10 Jahren durchgeführt. Streptococcus pneumoniae wurde bei 47 % der Patienten isoliert, Haemophilus influenzae bei 34 %, Moraxella catarrhalis bei 15 % und S. pyogenes bei 4 %.
Die Gesamtansprechrate von Streptococcus pneumoniae auf Cefixim war etwa 10 % niedriger und die von Haemophilus influenzae oder Moraxella catarrhalis etwa 7 % höher (12 %, wenn beta-Lactamase-positive Isolate von H. influenzae eingeschlossen sind) als die Ansprechraten dieser Organismen auf die aktiven Kontrollmedikamente.
In diesen Studien wurden die Patienten randomisiert und entweder mit Cefixim in Dosierungen von 4 mg/kg zweimal täglich oder 8 mg/kg einmal täglich oder mit einem Vergleichspräparat behandelt. Bei 69 bis 70 % der Patienten in jeder Gruppe verschwanden die Anzeichen und Symptome einer Mittelohrentzündung bei der Auswertung 2 bis 4 Wochen nach der Behandlung, aber bei 15 % der Patienten wurde ein anhaltender Erguss festgestellt. Bei der Auswertung am Ende der Therapie wurden 17 % der Patienten, die Cefixim erhielten, und 14 % der Patienten, die wirksame Vergleichsmedikamente erhielten (18 %, einschließlich der Patienten, die eine Haemophilus influenzae-Resistenz gegen das Kontrollmedikament hatten und die das antibakterielle Kontrollmedikament erhielten), als erkrankt angesehen Behandlungsfehler. Bei der 2- bis 4-wöchigen Nachbeobachtung hatten insgesamt 30 % bis 31 % der Patienten Anzeichen eines Behandlungsversagens oder eines Wiederauftretens der Erkrankung.
Bakteriologisches Ergebnis von Otitis media zwei bis vier Wochen nach der Therapie basierend auf wiederholtem Mittelohr
INFORMATIONEN ZUM PATIENTEN
Weisen Sie die Patienten darauf hin, dass antibakterielle Arzneimittel, einschließlich Cefixim, nur zur Behandlung bakterieller Infektionen eingesetzt werden sollten. Sie behandeln keine Virusinfektionen (z. B. Erkältung). Wenn Cefixim zur Behandlung einer bakteriellen Infektion verschrieben wird, sollten die Patienten darüber aufgeklärt werden, dass sie sich zwar zu Beginn der Therapie besser fühlen, das Medikament jedoch genau nach Anweisung eingenommen werden sollte. Das Auslassen von Dosen oder das Nichtbeenden des vollständigen Therapiezyklus kann: (1) die Wirksamkeit der sofortigen Behandlung verringern und (2) die Wahrscheinlichkeit erhöhen, dass Bakterien eine Resistenz entwickeln und nicht mit Cefixim zur Herstellung einer Suspension zum Einnehmen oder Cefixim-Kautabletten oder anderen behandelt werden können antibakterielle Medikamente in der Zukunft.
Weisen Sie Patienten mit Phenylketonurie darauf hin, dass SUPRAX 200 mg Kautabletten Aspartam, eine Phenylalaninquelle, wie folgt enthalten: Jede SUPRAX Kautablette enthält 3,3 mg, 5 mg und 6,7 mg Phenylalanin pro 100 mg, 150 mg bzw. 200 mg Stärke.
Weisen Sie die Patienten darauf hin, dass Durchfall ein häufiges Problem ist, das durch antibakterielle Medikamente verursacht wird und normalerweise endet, wenn das antibakterielle Medikament abgesetzt wird. Manchmal können Patienten nach Beginn der Behandlung mit antibakteriellen Arzneimitteln wässrigen und blutigen Stuhl (mit oder ohne Magenkrämpfe und Fieber) sogar noch zwei oder mehr Monate nach Einnahme der letzten Dosis des antibakteriellen Arzneimittels entwickeln. In diesem Fall sollten die Patienten so schnell wie möglich ihren Arzt kontaktieren.
Was ist Sustiva 600 mg und wie wird es angewendet?
Sustiva ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome einer HIV-Infektion. Sustiva kann allein oder mit anderen Medikamenten verwendet werden.
Sustiva gehört zu einer Klasse von Medikamenten namens HIV, NNRTIs.
Es ist nicht bekannt, ob Sustiva bei Kindern unter 3 Monaten sicher und wirksam ist.
Welche Nebenwirkungen kann Sustiva haben?
Sustiva kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Sustiva gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Sustiva. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
SUSTIVA® (Efavirenz) ist ein HIV-1-spezifischer, nicht-nukleosidischer Reverse-Transkriptase-Hemmer (NNRTI). Efavirenz wird chemisch als (S)-6-Chlor-4-(cyclopropylethinyl)-1,4-dihydro4-(trifluormethyl)-2H-3,1-benzoxazin-2-on beschrieben. Seine Summenformel ist C14H9ClF3NO2 und seine Strukturformel ist:
Efavirenz ist ein weißes bis leicht rosa kristallines Pulver mit einer Molekülmasse von 315,68. Es ist in Wasser praktisch unlöslich (
Kapseln
SUSTIVA ist als Kapseln zur oralen Verabreichung mit entweder 50 mg oder 200 mg Efavirenz und den folgenden Hilfsstoffen erhältlich: Lactose-Monohydrat, Magnesiumstearat, Natriumlaurylsulfat und Natriumstärkeglykolat. Die Kapselhülle enthält die folgenden Hilfsstoffe und Farbstoffe: Gelatine, Natriumlaurylsulfat, Titandioxid und/oder gelbes Eisenoxid. Die Kapselhüllen können auch Siliziumdioxid enthalten. Die Kapseln sind mit Tinte bedruckt, die Carmine 40 Blue, FD&C Blue No. 2 und Titandioxid enthält.
Tablets
SUSTIVA 600 mg ist als Filmtablette zur oralen Verabreichung mit 600 mg Efavirenz und den folgenden Hilfsstoffen erhältlich: Croscarmellose-Natrium, Hydroxypropylcellulose, Lactose-Monohydrat, Magnesiumstearat, mikrokristalline Cellulose und Natriumlaurylsulfat. Die Filmbeschichtung enthält Opadry Yellow und Opadry Clear. Die Tabletten sind mit Carnaubawachs poliert und mit violetter Tinte, Opacode WB, bedruckt.
INDIKATIONEN
SUSTIVA® (Efavirenz) in Kombination mit anderen antiretroviralen Wirkstoffen ist indiziert zur Behandlung einer Infektion mit dem humanen Immunschwächevirus Typ 1 (HIV-1) bei Erwachsenen und pädiatrischen Patienten ab einem Alter von 3 Monaten und einem Körpergewicht von mindestens 3,5 kg.
DOSIERUNG UND ANWENDUNG
Leberfunktion
Überwachen Sie die Leberfunktion vor und während der Behandlung mit SUSTIVA [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. SUSTIVA 600 mg wird bei Patienten mit mäßiger oder schwerer Leberfunktionsstörung (Child Pugh B oder C) nicht empfohlen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Erwachsene
Die empfohlene Dosierung von SUSTIVA (Efavirenz) beträgt 600 mg oral einmal täglich in Kombination mit einem Proteasehemmer und/oder nukleosidanalogen Reverse-Transkriptase-Hemmern (NRTIs). Es wird empfohlen, SUSTIVA auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Die nach Einnahme von SUSTIVA 600 mg mit Nahrung beobachteten erhöhten Efavirenz-Konzentrationen können zu einer Zunahme der Häufigkeit von Nebenwirkungen führen [siehe KLINISCHE PHARMAKOLOGIE ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit von Symptomen des Nervensystems verbessern [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , NEBENWIRKUNGEN , und INFORMATIONEN ZUM PATIENTEN ]. SUSTIVA Kapseln oder Tabletten sollten unzerkaut mit Flüssigkeit geschluckt werden. Für Patienten, die keine Kapseln oder Tabletten schlucken können, wird die Verabreichungsmethode mit Kapselspritzern empfohlen [siehe Kapsel besprühen Art der Verabreichung ].
Begleitende antiretrovirale Therapie
SUSTIVA muss in Kombination mit anderen antiretroviralen Medikamenten gegeben werden [siehe INDIKATIONEN , WARNUNGEN UND VORSICHTSMASSNAHMEN , WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN , und KLINISCHE PHARMAKOLOGIE ].
Dosisanpassung
Wenn SUSTIVA 200 mg zusammen mit Voriconazol angewendet wird, sollte die Voriconazol-Erhaltungsdosis auf 400 mg alle 12 Stunden erhöht und die SUSTIVA 200 mg-Dosis auf 300 mg einmal täglich unter Verwendung der Kapselformulierung (eine 200-mg- und zwei 50-mg-Kapseln oder sechs 50-mg-Kapseln) verringert werden mg-Kapseln). SUSTIVA 600 mg Tabletten dürfen nicht zerbrochen werden. [sehen WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE .]
Wenn SUSTIVA zusammen mit Rifampin an Patienten verabreicht wird, die 50 kg oder mehr wiegen, wird eine Erhöhung der Dosis von SUSTIVA 200 mg auf 800 mg einmal täglich empfohlen [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ].
Pädiatrische Patienten
Es wird empfohlen, SUSTIVA 600 mg auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Tabelle 1 beschreibt die empfohlene Dosis von SUSTIVA für pädiatrische Patienten im Alter von 3 Monaten oder älter und einem Gewicht zwischen 3,5 kg und 40 kg [siehe KLINISCHE PHARMAKOLOGIE ]. Die empfohlene Dosierung von SUSTIVA für pädiatrische Patienten mit einem Körpergewicht von 40 kg oder mehr beträgt 600 mg einmal täglich. Für pädiatrische Patienten, die keine Kapseln schlucken können, kann der Kapselinhalt mit einer kleinen Menge Nahrung oder Säuglingsanfangsnahrung unter Anwendung der Kapsel-Sprinkle-Methode verabreicht werden [siehe Kapsel besprühen Art der Verabreichung ].
Kapsel besprühen Art der Verabreichung
Bei pädiatrischen Patienten ab einem Alter von 3 Monaten und einem Gewicht von mindestens 3,5 kg sowie bei Erwachsenen, die Kapseln oder Tabletten nicht schlucken können, kann der Kapselinhalt mit einer kleinen Menge (1 bis 2 Teelöffel) Nahrung eingenommen werden. Die Verwendung von Säuglingsanfangsnahrung zum Mischen sollte nur für junge Säuglinge in Betracht gezogen werden, die feste Nahrung nicht zuverlässig zu sich nehmen können. Patienten und Pflegepersonal sollten angewiesen werden, die Kapsel vorsichtig zu öffnen, um ein Verschütten oder Verteilen des Kapselinhalts in die Luft zu vermeiden. Die Kapsel sollte waagerecht über einen kleinen Behälter gehalten und zum Öffnen vorsichtig gedreht werden. Bei Patienten, die feste Nahrung vertragen, sollte der gesamte Kapselinhalt in dem kleinen Behälter vorsichtig mit einer altersgerechten weichen Nahrung wie Apfelmus, Traubengelee oder Joghurt vermischt werden. Bei kleinen Säuglingen, die die Kapsel-Streusel-Säuglingsnahrung-Mischung erhalten, sollte der gesamte Kapselinhalt vorsichtig in 2 Teelöffel rekonstituierte Säuglingsnahrung bei Raumtemperatur in einem kleinen Behälter gemischt werden, indem vorsichtig mit einem kleinen Löffel umgerührt und die Mischung dann in eine 10 ml orale Dosierspritze zur Verabreichung. Nach der Verabreichung der SUSTIVA-Nahrungs- oder Nahrungsmischung muss eine zusätzliche kleine Menge (ca. 2 Teelöffel) Nahrung oder Nahrungsnahrung in den leeren Mischbehälter gegeben, gerührt werden, um alle verbleibenden SUSTIVA-Reste aufzulösen, und dem Patienten verabreicht werden. Die SUSTIVA-Nahrungs- oder Formelmischung sollte innerhalb von 30 Minuten nach dem Mischen verabreicht werden. Für 2 Stunden nach der Verabreichung von SUSTIVA sollte keine zusätzliche Nahrung aufgenommen werden.
Weitere Patientenhinweise zur Verabreichungsmethode mit Kapselstreuung finden sich in der von der FDA zugelassenen Patientenkennzeichnung (siehe INFORMATIONEN ZUM PATIENTEN und GEBRAUCHSANWEISUNG ).
WIE GELIEFERT
Darreichungsformen und Stärken
Kapseln
Die 200-mg-Kapseln sind goldfarben, mit dem Rückseitenaufdruck „SUSTIVA“ auf dem Unterteil und dem Aufdruck „200 mg“ auf dem Oberteil.
Die 50-mg-Kapseln sind goldfarben und weiß, mit dem Aufdruck „SUSTIVA“ auf dem goldfarbenen Oberteil und dem umgekehrten Aufdruck „50 mg“ auf dem weißen Unterteil.
Tablets
600-mg-Tabletten sind gelbe, kapselförmige Filmtabletten mit dem Aufdruck „SUSTIVA“ auf beiden Seiten.
Lagerung und Handhabung
Kapseln
SUSTIVA® (Efavirenz) Kapseln sind wie folgt erhältlich:
Die 200-mg-Kapseln sind goldfarben, mit dem Rückseitenaufdruck „SUSTIVA“ auf dem Unterteil und dem Aufdruck „200 mg“ auf dem Oberteil.
Flaschen von 90 - NDC 0056-0474-92
Die 50-mg-Kapseln sind goldfarben und weiß, mit dem Aufdruck „SUSTIVA“ auf dem goldfarbenen Oberteil und dem umgekehrten Aufdruck „50 mg“ auf dem weißen Unterteil.
Flaschen von 30 - NDC 0056-0470-30
Tablets
SUSTIVA® (Efavirenz) Tabletten sind wie folgt erhältlich:
Tabletten 600 mg sind gelbe, kapselförmige Filmtabletten mit dem Aufdruck „SUSTIVA“ auf beiden Seiten.
Flaschen von 30 - NDC 0056-0510-30
Lagerung
SUSTIVA 600 mg Kapseln und SUSTIVA Tabletten sollten bei 25 °C (77 °F) gelagert werden; Abweichungen bis 15°C-30°C (59°F-86°F) zulässig [siehe USP Controlled Room Temperature].
Vertrieb durch: Bristol-Myers Squibb Company Princeton, NJ 08543 USA. Überarbeitet: Okt. 2020
NEBENWIRKUNGEN
Die wichtigsten Nebenwirkungen, die bei mit SUSTIVA behandelten Patienten beobachtet wurden, sind:
Erfahrung mit klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die berichteten Nebenwirkungsraten nicht direkt mit den Raten in anderen klinischen Studien verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Nebenwirkungen bei Erwachsenen
Die häufigsten (> 5 % in beiden Efavirenz-Behandlungsgruppen) Nebenwirkungen von mindestens mäßigem Schweregrad bei Patienten in Studie 006, die mit SUSTIVA 200 mg in Kombination mit Zidovudin/Lamivudin oder Indinavir behandelt wurden, waren Hautausschlag, Schwindel, Übelkeit, Kopfschmerzen, Müdigkeit, Schlaflosigkeit, und Erbrechen.
Ausgewählte klinische Nebenwirkungen mittlerer oder schwerer Intensität, die bei ≥ 2 % der mit SUSTIVA behandelten Patienten in zwei kontrollierten klinischen Studien beobachtet wurden, sind in Tabelle 2 aufgeführt.
Pankreatitis wurde berichtet, obwohl ein kausaler Zusammenhang mit Efavirenz nicht nachgewiesen wurde. Asymptomatische Anstiege der Amylasespiegel im Serum wurden bei einer signifikant höheren Anzahl von mit Efavirenz 600 mg behandelten Patienten beobachtet als bei Kontrollpatienten (vgl Laboranomalien ).
Symptome des Nervensystems
Für 1008 Patienten, die mit SUSTIVA-haltigen Regimen behandelt wurden, und 635 Patienten, die in kontrollierten Studien mit einem Kontrollregime behandelt wurden, listet Tabelle 3 die Häufigkeit von Symptomen unterschiedlicher Schweregrade auf und gibt die Abbruchraten für eines oder mehrere der folgenden Symptome des Nervensystems an: Schwindel, Schlaflosigkeit, Konzentrationsstörungen, Somnolenz, anormales Träumen, Euphorie, Verwirrtheit, Erregung, Amnesie, Halluzinationen, Benommenheit, anormales Denken und Depersonalisation [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Häufigkeiten spezifischer Symptome des zentralen und peripheren Nervensystems sind in Tabelle 2 angegeben.
Psychiatrische Symptome
Bei mit SUSTIVA behandelten Patienten wurde über schwerwiegende psychiatrische Nebenwirkungen berichtet. In kontrollierten Studien waren Depression (19 %, 16 %), Angst (13 %, 9 %) und Nervosität (7 %) psychiatrische Symptome, die mit einer Häufigkeit von mehr als 2 % bei mit SUSTIVA 600 mg bzw. Kontrollregimen behandelten Patienten beobachtet wurden. , 2 %).
Ausschlag
In kontrollierten klinischen Studien betrug die Häufigkeit von Hautausschlägen (alle Schweregrade, unabhängig von der Kausalität) 26 % bei 1008 Erwachsenen, die mit Therapien behandelt wurden, die SUSTIVA 200 mg enthielten, und 17 % bei 635 Erwachsenen, die mit einer Kontrolltherapie behandelt wurden. Die meisten Berichte über Hautausschlag waren leicht oder mittelschwer. Die Häufigkeit von Hautausschlag Grad 3 betrug 0,8 % bei mit SUSTIVA behandelten Patienten und 0,3 % bei den Kontrollgruppen, und die Häufigkeit von Hautausschlag Grad 4 betrug 0,1 % bei SUSTIVA und 0 bei Kontrollgruppen. Die Abbruchraten aufgrund von Hautausschlag betrugen 1,7 % bei den mit SUSTIVA behandelten Patienten und 0,3 % bei den Kontrollgruppen [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Die Erfahrungen mit SUSTIVA bei Patienten, die andere antiretrovirale Arzneimittel der NNRTI-Klasse abgesetzt haben, sind begrenzt. Neunzehn Patienten, die Nevirapin wegen Hautausschlag abgesetzt hatten, wurden mit SUSTIVA behandelt. Neun dieser Patienten entwickelten während der Therapie mit SUSTIVA einen leichten bis mittelschweren Hautausschlag, und zwei dieser Patienten brachen die Behandlung wegen Hautausschlag ab.
Laboranomalien
Ausgewählte Laboranomalien der Grade 3-4, die bei ≥ 2 % der mit SUSTIVA behandelten Patienten in zwei klinischen Studien berichtet wurden, sind in Tabelle 4 aufgeführt.
Patienten, die gleichzeitig mit Hepatitis B oder C infiziert sind
Leberfunktionstests sollten bei Patienten mit Hepatitis B und/oder C in der Anamnese überwacht werden. Im Langzeitdatensatz von Studie 006 wurden 137 Patienten mit SUSTIVA-haltigen Regimen behandelt (mittlere Therapiedauer 68 Wochen) und 84 behandelt mit einem Kontrollregime (mittlere Dauer 56 Wochen) waren beim Screening auf Hepatitis B (Oberflächenantigen-positiv) und/oder C (Hepatitis-C-Antikörper-positiv) seropositiv. Unter diesen koinfizierten Patienten traten bei 13 % der Patienten in den SUSTIVA-Armen und 7 % der Patienten im Kontrollarm Erhöhungen der AST auf mehr als das Fünffache der ULN auf, und bei 20 % der Patienten traten Erhöhungen der ALT auf mehr als das Fünffache der ULN auf in den SUSTIVA-Armen und 7 % der Patienten im Kontrollarm. Unter den koinfizierten Patienten brachen 3 % der mit SUSTIVA-haltigen Regimen behandelten und 2 % im Kontrollarm die Studie wegen Leber- oder Gallenwegserkrankungen ab [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Lipide
Bei einigen nicht infizierten Probanden, die SUSTIVA erhielten, wurde ein Anstieg des Gesamtcholesterins gegenüber dem Ausgangswert um 10-20 % beobachtet. Bei Patienten, die mit SUSTIVA + Zidovudin + Lamivudin behandelt wurden, wurden Anstiege von ungefähr 20 % bzw. 25 % gegenüber dem Ausgangswert von Gesamtcholesterin und HDL ohne Nüchternblut beobachtet. Bei Patienten, die mit SUSTIVA + Indinavir behandelt wurden, wurden Anstiege von Cholesterin und HDL außerhalb des Nüchternzustands um etwa 40 % bzw. 35 % gegenüber dem Ausgangswert beobachtet. Gesamtcholesterinspiegel außerhalb des Nüchternzustands von ≥ 240 mg/dl und ≥ 300 mg/dl wurden bei 34 % bzw. 9 % der mit SUSTIVA + Zidovudin + Lamivudin behandelten Patienten berichtet; 54 % bzw. 20 % der mit SUSTIVA + Indinavir behandelten Patienten; und 28 % bzw. 4 % der mit Indinavir + Zidovudin + Lamivudin behandelten Patienten. Die Wirkungen von SUSTIVA 600 mg auf Triglyceride und LDL in dieser Studie waren nicht gut charakterisiert, da die Proben von nicht nüchternen Patienten genommen wurden. Die klinische Bedeutung dieser Befunde ist unbekannt [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Nebenwirkungen bei pädiatrischen Patienten
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die berichteten Nebenwirkungsraten nicht direkt mit den Raten in anderen klinischen Studien verglichen werden und spiegeln möglicherweise nicht die in der klinischen Praxis beobachteten Raten wider.
Die Bewertung der Nebenwirkungen basiert auf drei klinischen Studien mit 182 HIV-1-infizierten pädiatrischen Patienten (im Alter von 3 Monaten bis 21 Jahren), die SUSTIVA 200 mg in Kombination mit anderen antiretroviralen Wirkstoffen über einen medianen Zeitraum von 123 Wochen erhielten. Die in den drei Studien beobachteten Nebenwirkungen ähnelten denen, die in klinischen Studien bei Erwachsenen beobachtet wurden, mit der Ausnahme, dass Hautausschlag häufiger bei pädiatrischen Patienten auftrat (32 % für alle Schweregrade, unabhängig von der Kausalität) und häufiger von höherem Schweregrad (dh schwerer). Bei zwei (1,1 %) pädiatrischen Patienten kam es zu einem Hautausschlag Grad 3 (konfluierender Hautausschlag mit Fieber, generalisierter Hautausschlag) und bei vier (2,2 %) pädiatrischen Patienten kam es zu einem Hautausschlag Grad 4 (alles Erythema multiforme). Fünf pädiatrische Patienten (2,7 %) brachen die Studie wegen Hautausschlag ab [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von SUSTIVA nach der Zulassung festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet wurden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen.
Körper als Ganzes: allergische Reaktionen, Asthenie, Umverteilung/Ansammlung von Körperfett [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]
Zentrales und peripheres Nervensystem: Koordinationsstörungen, Ataxie, Enzephalopathie, zerebelläre Koordinations- und Gleichgewichtsstörungen, Krämpfe, Hypästhesie, Parästhesien, Neuropathie, Tremor, Schwindel
Endokrin: Gynäkomastie
Magen-Darm: Verstopfung, Malabsorption
Herz-Kreislauf: Erröten, Herzklopfen
Leber und Gallensystem: Anstieg der Leberenzyme, Leberversagen, Hepatitis.
Stoffwechsel und Ernährung: Hypercholesterinämie, Hypertriglyzeridämie
Bewegungsapparat: Arthralgie, Myalgie, Myopathie
Psychiatrie: aggressive Reaktionen, Erregung, Wahnvorstellungen, emotionale Labilität, Manie, Neurose, Paranoia, Psychose, Selbstmord, Katatonie
Atmung: Dyspnoe
Haut und Anhängsel: Erythema multiforme, photoallergische Dermatitis, Stevens-Johnson-Syndrom
Besondere Sinne: Sehstörungen, Tinnitus
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Mögliche Auswirkungen von SUSTIVA auf andere Medikamente
Es wurde gezeigt, dass Efavirenz in vivo CYP3A und CYP2B6 induziert. Andere Verbindungen, die Substrate von CYP3A oder CYP2B6 sind, können verringerte Plasmakonzentrationen aufweisen, wenn sie zusammen mit SUSTIVA verabreicht werden.
Mögliche Beeinflussung von SUSTIVA durch andere Medikamente
Es ist zu erwarten, dass Arzneimittel, die eine CYP3A-Aktivität induzieren (z. B. Phenobarbital, Rifampin, Rifabutin), die Clearance von Efavirenz erhöhen, was zu verringerten Plasmakonzentrationen führt [siehe DOSIERUNG UND ANWENDUNG ].
QT-verlängernde Medikamente
Es liegen nur begrenzte Informationen über die Möglichkeit einer pharmakodynamischen Wechselwirkung zwischen SUSTIVA 600 mg und Arzneimitteln vor, die das QTc-Intervall verlängern. Bei Anwendung von Efavirenz wurde eine QTc-Verlängerung beobachtet [siehe KLINISCHE PHARMAKOLOGIE ]. Ziehen Sie Alternativen zu SUSTIVA 600 mg in Betracht, wenn es zusammen mit einem Medikament verabreicht wird, bei dem ein bekanntes Torsade-de-Pointes-Risiko besteht.
Gegründete und andere potenziell signifikante Wechselwirkungen mit anderen Arzneimitteln
Arzneimittelwechselwirkungen mit SUSTIVA sind in Tabelle 5 zusammengefasst. Für Daten zur Pharmakokinetik [siehe KLINISCHE PHARMAKOLOGIE ] Tabellen 7 und 8. Diese Tabelle enthält potenziell signifikante Wechselwirkungen, ist jedoch nicht vollständig.
Arzneimittel ohne klinisch signifikante Wechselwirkungen mit SUSTIVA
Es wird keine Dosisanpassung empfohlen, wenn SUSTIVA 600 mg zusammen mit den folgenden Arzneimitteln gegeben wird: Aluminium-/Magnesiumhydroxid-Antazida, Azithromycin, Cetirizin, Famotidin, Fluconazol, Lorazepam, Nelfinavir, nukleosidische Reverse-Transkriptase-Hemmer (Abacavir, Emtricitabin, Lamivudin, Stavudin, Tenofovirdisoproxilfumarat, Zidovudin). ), Paroxetin und Raltegravir.
Cannabinoid-Test-Interaktion
Efavirenz bindet nicht an Cannabinoidrezeptoren. Bei einigen Screening-Assays bei nicht infizierten und HIV-infizierten Probanden, die Efavirenz erhielten, wurde über falsch-positive Cannabinoid-Testergebnisse im Urin berichtet. Die Bestätigung positiver Screening-Tests für Cannabinoide durch eine spezifischere Methode wird empfohlen.
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Wechselwirkungen mit anderen Medikamenten
Die Plasmakonzentrationen von Efavirenz können durch Substrate, Inhibitoren oder Induktoren von CYP3A verändert werden. Ebenso kann Efavirenz die Plasmakonzentrationen von Arzneimitteln verändern, die durch CYP3A oder CYP2B6 metabolisiert werden. Die hervorstechendste Wirkung von Efavirenz im Steady State ist die Induktion von CYP3A und CYP2B6 [vgl DOSIERUNG UND ANWENDUNG und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
QTc-Verlängerung
Bei Anwendung von Efavirenz wurde eine QTc-Verlängerung beobachtet [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ]. Ziehen Sie Alternativen zu SUSTIVA 600 mg in Betracht, wenn es zusammen mit einem Arzneimittel verabreicht wird, bei dem ein bekanntes Risiko für Torsade de Pointes besteht, oder wenn es Patienten mit einem höheren Risiko für Torsade de Pointes verabreicht wird.
Widerstand
SUSTIVA 200 mg darf nicht als Einzelwirkstoff zur Behandlung einer HIV-1-Infektion angewendet oder als Einzelwirkstoff zu einem versagenden Regime hinzugefügt werden. Resistente Viren treten schnell auf, wenn Efavirenz als Monotherapie verabreicht wird. Bei der Auswahl neuer antiretroviraler Wirkstoffe, die in Kombination mit Efavirenz angewendet werden sollen, sollte das Potenzial für eine virale Kreuzresistenz berücksichtigt werden.
Koadministration mit verwandten Produkten
Die gleichzeitige Anwendung von SUSTIVA mit ATRIPLA (Efavirenz 600 mg/Emtricitabin 200 mg/Tenofovirdisoproxilfumarat 300 mg) wird nicht empfohlen, es sei denn, dies ist zur Dosisanpassung erforderlich (z. B. mit Rifampin), da Efavirenz einer seiner Wirkstoffe ist.
Psychiatrische Symptome
Bei mit SUSTIVA behandelten Patienten wurde über schwerwiegende psychiatrische Nebenwirkungen berichtet. In kontrollierten Studien mit 1008 Patienten, die durchschnittlich 2,1 Jahre lang mit 600 mg SUSTIVA-Regimen behandelt wurden, und 635 Patienten, die durchschnittlich 1,5 Jahre lang mit Kontrollschemata behandelt wurden, wurde die Häufigkeit (unabhängig von der Kausalität) spezifischer schwerwiegender psychiatrischer Ereignisse bei Patienten, die SUSTIVA erhielten, oder Kontrollschemata waren schwere Depressionen (2,4 %, 0,9 %), Selbstmordgedanken (0,7 %, 0,3 %), nicht tödliche Selbstmordversuche (0,5 %, 0), aggressives Verhalten (0,4 %, 0,5 %), paranoide Reaktionen (0,4 %, 0,3 %) und manische Reaktionen (0,2 %, 0,3 %). Wenn ähnliche psychiatrische Symptome wie die oben genannten kombiniert und in einer multifaktoriellen Analyse der Daten aus Studie 006 als Gruppe ausgewertet wurden, war die Behandlung mit Efavirenz mit einem vermehrten Auftreten dieser ausgewählten psychiatrischen Symptome verbunden. Andere Faktoren, die mit einem Anstieg des Auftretens dieser psychiatrischen Symptome verbunden sind, waren die Vorgeschichte des intravenösen Drogenkonsums, die psychiatrische Vorgeschichte und die Einnahme von psychiatrischen Medikamenten bei Studieneintritt; ähnliche Assoziationen wurden sowohl in der SUSTIVA- als auch in der Kontrollgruppe beobachtet. In Studie 006 traten während der gesamten Studie sowohl bei den mit SUSTIVA behandelten als auch bei den Kontrollpatienten neue schwerwiegende psychiatrische Symptome auf. Ein Prozent der mit SUSTIVA behandelten Patienten hat die Behandlung wegen eines oder mehrerer dieser ausgewählten psychiatrischen Symptome abgebrochen oder unterbrochen. Es gab auch gelegentliche Postmarketing-Berichte über Tod durch Suizid, Wahnvorstellungen und psychoseähnliches Verhalten, obwohl aus diesen Berichten kein kausaler Zusammenhang mit der Anwendung von SUSTIVA ermittelt werden kann. Postmarketing-Fälle von Katatonie wurden ebenfalls berichtet und können mit einer erhöhten Efavirenz-Exposition in Zusammenhang stehen. Patienten mit schwerwiegenden psychiatrischen Nebenwirkungen sollten sich umgehend medizinisch untersuchen lassen, um die Möglichkeit abzuschätzen, dass die Symptome mit der Anwendung von SUSTIVA in Zusammenhang stehen könnten, und falls ja, um festzustellen, ob die Risiken einer fortgesetzten Therapie den Nutzen überwiegen. [sehen NEBENWIRKUNGEN .]
Symptome des Nervensystems
Dreiundfünfzig Prozent (531/1008) der Patienten, die SUSTIVA 600 mg in kontrollierten Studien erhielten, berichteten über Symptome des Zentralnervensystems (jeden Grades, unabhängig von der Kausalität), verglichen mit 25 % (156/635) der Patienten, die Kontrollregime erhielten [siehe NEBENWIRKUNGEN ]. Zu diesen Symptomen gehörten unter anderem Schwindel (28,1 % der 1008 Patienten), Schlaflosigkeit (16,3 %), Konzentrationsstörungen (8,3 %), Somnolenz (7,0 %), abnormale Träume (6,2 %) und Halluzinationen (1.2 %). Diese Symptome waren bei 2,0 % der Patienten schwerwiegend; und 2,1 % der Patienten brachen daraufhin die Therapie ab. Diese Symptome beginnen normalerweise am ersten oder zweiten Therapietag und klingen im Allgemeinen nach den ersten 2-4 Wochen der Therapie ab. Nach 4-wöchiger Therapie lag die Prävalenz von Symptomen des Nervensystems von mindestens mäßigem Schweregrad zwischen 5 % und 9 % bei Patienten, die mit SUSTIVA-haltigen Regimen behandelt wurden, und zwischen 3 % und 5 % bei Patienten, die mit einem Kontrollregime behandelt wurden. Die Patienten sollten darüber informiert werden, dass sich diese häufigen Symptome bei Fortführung der Therapie wahrscheinlich bessern und nicht vorhersagend für das spätere Auftreten der weniger häufigen psychiatrischen Symptome sind [siehe Psychiatrische Symptome ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit dieser Symptome des Nervensystems verbessern [siehe DOSIERUNG UND ANWENDUNG ].
Die Analyse der Langzeitdaten aus Studie 006 (mediane Nachbeobachtungszeit 180 Wochen, 102 Wochen bzw. 76 Wochen für Patienten, die mit SUSTIVA + Zidovudin + Lamivudin, SUSTIVA + Indinavir bzw. Indinavir + Zidovudin + Lamivudin behandelt wurden) zeigte dies darüber hinaus Nach 24-wöchiger Therapie war die Inzidenz neu aufgetretener Symptome des Nervensystems bei den mit SUSTIVA behandelten Patienten im Allgemeinen ähnlich wie im Indinavir-enthaltenden Kontrollarm.
Eine spät einsetzende Neurotoxizität, einschließlich Ataxie und Enzephalopathie (Bewusstseinsstörungen, Verwirrtheit, psychomotorische Verlangsamung, Psychose, Delirium), kann Monate bis Jahre nach Beginn der Efavirenz-Therapie auftreten. Bei Patienten mit genetischen CYP2B6-Polymorphismen, die trotz Standarddosierung von SUSTIVA mit erhöhten Efavirenz-Spiegeln einhergehen, traten einige Ereignisse mit spät einsetzender Neurotoxizität auf. Patienten mit Anzeichen und Symptomen schwerwiegender neurologischer Nebenwirkungen sollten umgehend untersucht werden, um die Möglichkeit zu beurteilen, dass diese Ereignisse mit der Anwendung von Efavirenz in Zusammenhang stehen und ob ein Absetzen von SUSTIVA 600 mg gerechtfertigt ist.
Patienten, die SUSTIVA 600 mg erhalten, sollten auf die Möglichkeit zusätzlicher Wirkungen auf das Zentralnervensystem aufmerksam gemacht werden, wenn SUSTIVA 200 mg gleichzeitig mit Alkohol oder Psychopharmaka angewendet wird.
Patienten mit zentralnervösen Symptomen wie Schwindel, Konzentrationsstörungen und/oder Schläfrigkeit sollten potenziell gefährliche Tätigkeiten wie Autofahren oder Bedienen von Maschinen vermeiden.
Embryo-fötale Toxizität
Efavirenz kann den Fötus schädigen, wenn es einer schwangeren Frau während des ersten Trimenons verabreicht wird. Weisen Sie gebärfähige Frauen, die SUSTIVA 600 mg erhalten, darauf hin, eine Schwangerschaft zu vermeiden. [sehen Verwendung in bestimmten Bevölkerungsgruppen .]
Ausschlag
In kontrollierten klinischen Studien traten bei 26 % (266/1008) der erwachsenen Patienten, die mit 600 mg SUSTIVA behandelt wurden, neue Hautausschläge auf, verglichen mit 17 % (111/635) der in den Kontrollgruppen behandelten Patienten [siehe NEBENWIRKUNGEN ]. Bei 0,9 % (9/1008) der mit SUSTIVA behandelten Patienten trat ein mit Blasenbildung, feuchter Abschuppung oder Ulzeration einhergehender Hautausschlag auf. Die Inzidenz von Ausschlag 4. Grades (z. B. Erythema multiforme, Stevens-Johnson-Syndrom) bei erwachsenen Patienten, die mit SUSTIVA in allen Studien und erweitertem Zugang behandelt wurden, betrug 0,1 %. Hautausschläge sind in der Regel leichte bis mittelschwere makulopapulöse Hautausschläge, die innerhalb der ersten 2 Wochen nach Beginn der Therapie mit Efavirenz auftreten (die mediane Zeit bis zum Auftreten des Hautausschlags bei Erwachsenen betrug 11 Tage) und bei den meisten Patienten, die die Therapie mit Efavirenz fortsetzen, verschwindet der Hautausschlag innerhalb von 1 Tag Monat (mittlere Dauer 16 Tage). Die Abbruchrate wegen Hautausschlag in klinischen Studien mit Erwachsenen betrug 1,7 % (17/1008).
Ausschlag wurde bei 59 von 182 pädiatrischen Patienten (32 %), die mit SUSTIVA behandelt wurden, berichtet [siehe NEBENWIRKUNGEN ]. Bei zwei pädiatrischen Patienten kam es zu einem Hautausschlag Grad 3 (konfluierender Hautausschlag mit Fieber, generalisierter Hautausschlag), und vier Patienten hatten einen Hautausschlag Grad 4 (Erythema multiforme). Die mediane Zeit bis zum Auftreten des Hautausschlags bei pädiatrischen Patienten betrug 28 Tage (Bereich 3–1642 Tage). Vor Beginn der Therapie mit SUSTIVA bei pädiatrischen Patienten sollte eine Prophylaxe mit geeigneten Antihistaminika in Betracht gezogen werden.
SUSTIVA 600 mg kann im Allgemeinen bei Patienten wieder aufgenommen werden, die die Therapie aufgrund eines Hautausschlags unterbrechen. SUSTIVA 200 mg sollte bei Patienten abgesetzt werden, die einen schweren Hautausschlag entwickeln, der mit Blasenbildung, Abschuppung, Schleimhautbeteiligung oder Fieber einhergeht. Geeignete Antihistaminika und/oder Kortikosteroide können die Verträglichkeit verbessern und das Abklingen des Hautausschlags beschleunigen. Bei Patienten, die eine lebensbedrohliche Hautreaktion (z. B. Stevens-Johnson-Syndrom) hatten, sollte eine alternative Therapie in Betracht gezogen werden [siehe KONTRAINDIKATIONEN ].
Hepatotoxizität
Postmarketing-Fälle von Hepatitis, einschließlich fulminanter Hepatitis, die zu einem Leberversagen fortschreiten, das eine Transplantation erfordert oder zum Tod führt, wurden bei mit SUSTIVA behandelten Patienten berichtet. Die Berichte umfassten Patienten mit zugrunde liegender Lebererkrankung, einschließlich Koinfektion mit Hepatitis B oder C, und Patienten ohne vorbestehende Lebererkrankung oder andere identifizierbare Risikofaktoren.
SUSTIVA 600 mg wird für Patienten mit mäßiger oder schwerer Leberfunktionsstörung nicht empfohlen. Bei Patienten mit leichter Leberfunktionsstörung, die SUSTIVA erhalten, wird eine sorgfältige Überwachung empfohlen. [sehen NEBENWIRKUNGEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Eine Überwachung der Leberenzyme vor und während der Behandlung wird für alle Patienten empfohlen [siehe DOSIERUNG UND ANWENDUNG ].
Erwägen Sie das Absetzen von SUSTIVA 600 mg bei Patienten mit anhaltenden Erhöhungen der Serumtransaminasen auf mehr als das Fünffache der Obergrenze des Normalbereichs.Setzen Sie SUSTIVA 200 mg ab, wenn die Erhöhung der Serumtransaminasen von klinischen Anzeichen oder Symptomen einer Hepatitis oder Leberdekompensation begleitet wird.
Krämpfe
Bei erwachsenen und pädiatrischen Patienten, die Efavirenz erhielten, wurden Konvulsionen beobachtet, im Allgemeinen bei Vorliegen bekannter Anfälle in der Krankengeschichte [siehe Nichtklinische Toxikologie ]. Vorsicht ist geboten bei allen Patienten mit Krampfanfällen in der Anamnese. Bei Patienten, die gleichzeitig Antikonvulsiva erhalten, die hauptsächlich von der Leber metabolisiert werden, wie Phenytoin und Phenobarbital, kann eine regelmäßige Überwachung der Plasmaspiegel erforderlich sein [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Lipiderhöhungen
Die Behandlung mit SUSTIVA hat zu einem Anstieg der Konzentration von Gesamtcholesterin und Triglyceriden geführt [siehe NEBENWIRKUNGEN ]. Cholesterin- und Triglyceridtests sollten vor Beginn der Therapie mit SUSTIVA 200 mg und in regelmäßigen Abständen während der Therapie durchgeführt werden.
Immunrekonstitutionssyndrom
Bei Patienten, die mit einer antiretroviralen Kombinationstherapie, einschließlich SUSTIVA, behandelt wurden, wurde über ein Immunrekonstitutionssyndrom berichtet. Während der Anfangsphase einer antiretroviralen Kombinationsbehandlung können Patienten, deren Immunsystem anspricht, eine entzündliche Reaktion auf indolente oder verbleibende opportunistische Infektionen (wie Mycobacterium avium-Infektion, Cytomegalovirus, Pneumocystis-jiroveci-Pneumonie [PCP] oder Tuberkulose) entwickeln, die eine weitere Untersuchung erforderlich machen kann und Behandlung.
Es wurde auch über das Auftreten von Autoimmunerkrankungen (wie Morbus Basedow, Polymyositis, Guillain-Barré-Syndrom und Autoimmunhepatitis) im Zusammenhang mit der Immunrekonstitution berichtet; Die Zeit bis zum Einsetzen ist jedoch variabler und kann viele Monate nach Beginn der Behandlung auftreten.
Fettumverteilung
Bei Patienten, die eine antiretrovirale Therapie erhielten, wurde eine Umverteilung/Ansammlung von Körperfett einschließlich zentraler Adipositas, dorsozervikaler Fettvergrößerung (Büffelbuckel), peripherer Auszehrung, Gesichtsauszehrung, Brustvergrößerung und „cushingoidem Aussehen“ beobachtet. Der Mechanismus und die langfristigen Folgen dieser Ereignisse sind derzeit unbekannt. Ein kausaler Zusammenhang wurde nicht hergestellt.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( INFORMATIONEN ZUM PATIENTEN und Gebrauchsanweisung ).
Wechselwirkungen mit anderen Medikamenten
Auf den Flaschenetiketten des Produkts befindet sich eine Erklärung für Patienten und medizinisches Fachpersonal: ACHTUNG: Informieren Sie sich über Arzneimittel, die NICHT zusammen mit SUSTIVA eingenommen werden sollten.
SUSTIVA kann mit einigen Arzneimitteln interagieren; raten Sie den Patienten daher, die Verwendung anderer verschreibungspflichtiger oder nicht verschreibungspflichtiger Medikamente ihrem Arzt zu melden.
Allgemeine Informationen für Patienten
Informieren Sie die Patienten darüber, dass SUSTIVA 200 mg keine Heilung für eine HIV-1-Infektion darstellt und die Patienten möglicherweise weiterhin an Krankheiten im Zusammenhang mit einer HIV-1-Infektion leiden, einschließlich opportunistischer Infektionen. Die Patienten sollten während der Einnahme von SUSTIVA in ärztlicher Behandlung bleiben.
Raten Sie den Patienten, Dinge zu vermeiden, die eine HIV-1-Infektion auf andere übertragen können.
Dosierungsanleitung
Empfehlen Sie den Patienten, SUSTIVA jeden Tag wie verordnet einzunehmen. Wenn ein Patient die Einnahme von SUSTIVA vergisst, sagen Sie ihm, dass er die vergessene Dosis sofort einnehmen soll, es sei denn, es ist fast Zeit für die nächste Dosis. Weisen Sie den Patienten darauf hin, nicht 2 Dosen auf einmal einzunehmen und die nächste Dosis zum regulär geplanten Zeitpunkt einzunehmen. Raten Sie dem Patienten, einen Arzt zu fragen, ob er/sie Hilfe bei der Planung der besten Zeiten für die Einnahme seiner/ihrer Medikamente benötigt.
SUSTIVA 200 mg muss immer in Kombination mit anderen antiretroviralen Arzneimitteln angewendet werden. Empfehlen Sie den Patienten, SUSTIVA 600 mg auf nüchternen Magen einzunehmen, vorzugsweise vor dem Schlafengehen. Die Einnahme von SUSTIVA zusammen mit Nahrung erhöht die Konzentration von Efavirenz und kann die Häufigkeit von Nebenwirkungen erhöhen. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit von Symptomen des Nervensystems verbessern [siehe DOSIERUNG UND ANWENDUNG und NEBENWIRKUNGEN ]. Gesundheitsdienstleister sollten Eltern oder Betreuungspersonen bei der Bestimmung des besten SUSTIVA-Dosierungsplans für Säuglinge und Kleinkinder unterstützen.
Für erwachsene und pädiatrische Patienten, die keine Kapseln oder Tabletten schlucken können, sollten Patienten oder ihre Betreuungspersonen angewiesen werden, die Anweisungen zur Verabreichung des Kapselinhalts in einer kleinen Menge Nahrung oder Säuglingsanfangsnahrung zu lesen und sorgfältig zu befolgen [siehe DOSIERUNG UND ANWENDUNG und FDA-zugelassene Patientenkennzeichnung ( INFORMATIONEN ZUM PATIENTEN und GEBRAUCHSANWEISUNG )]. Patienten sollten ihren Arzt oder Apotheker anrufen, wenn sie Fragen haben.
Symptome des Nervensystems
Informieren Sie die Patienten darüber, dass Symptome des Zentralnervensystems (NSS), einschließlich Schwindel, Schlaflosigkeit, Konzentrationsstörungen, Benommenheit und abnormale Träume, häufig während der ersten Wochen der Therapie mit SUSTIVA berichtet werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Die Einnahme vor dem Schlafengehen kann die Verträglichkeit dieser Symptome verbessern, die sich wahrscheinlich bei fortgesetzter Therapie bessern werden. Machen Sie die Patienten auf mögliche additive Wirkungen aufmerksam, wenn SUSTIVA gleichzeitig mit Alkohol oder Psychopharmaka angewendet wird. Weisen Sie die Patienten darauf hin, dass sie potenziell gefährliche Tätigkeiten wie Autofahren oder Bedienen von Maschinen vermeiden sollten, wenn sie NSS erleben.
Informieren Sie die Patienten darüber, dass ein Risiko für die Entwicklung einer spät einsetzenden Neurotoxizität besteht, einschließlich Ataxie und Enzephalopathie, die Monate bis Jahre nach Beginn der SUSTIVA-Therapie auftreten können [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Psychiatrische Symptome
Informieren Sie die Patienten darüber, dass bei Patienten, die SUSTIVA erhielten, über schwerwiegende psychiatrische Symptome einschließlich schwerer Depression, Suizidversuche, aggressives Verhalten, Wahnvorstellungen, Paranoia, psychoseähnliche Symptome und Katatonie berichtet wurde [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Wenn sie schwerwiegende psychiatrische Nebenwirkungen erfahren, sollten sie sich unverzüglich medizinisch untersuchen lassen. Raten Sie den Patienten, ihren Arzt über jede Vorgeschichte von psychischen Erkrankungen oder Drogenmissbrauch zu informieren.
Ausschlag
Informieren Sie die Patienten darüber, dass Hautausschlag eine häufige Nebenwirkung ist [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Hautausschläge verschwinden in der Regel ohne Änderung der Behandlung. Da der Hautausschlag jedoch schwerwiegend sein kann, raten Sie den Patienten, sich unverzüglich an ihren Arzt zu wenden, wenn ein Hautausschlag auftritt.
Hepatotoxizität
Weisen Sie die Patienten darauf hin, auf frühe Warnzeichen einer Leberentzündung oder eines Leberversagens, wie Müdigkeit, Schwäche, Appetitlosigkeit, Übelkeit und Erbrechen, sowie auf spätere Anzeichen wie Gelbsucht, Verwirrtheit, Bauchschwellung und verfärbten Kot zu achten und ihren Arzt zu konsultieren unverzüglich einen Arzt aufsuchen, wenn solche Symptome auftreten [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Weibchen mit reproduktivem Potenzial
Raten Sie gebärfähigen Frauen, während der Behandlung mit SUSTIVA 600 mg und für 12 Wochen nach Absetzen von SUSTIVA eine wirksame Verhütungsmethode sowie eine Barrieremethode anzuwenden. Weisen Sie Patientinnen darauf hin, sich an ihren Arzt zu wenden, wenn sie eine Schwangerschaft planen, schwanger werden oder wenn während der Behandlung mit SUSTIVA eine Schwangerschaft vermutet wird [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Schwangerschafts-Expositionsregister
Weisen Sie die Patientinnen darauf hin, dass es ein Schwangerschaftsexpositionsregister gibt, das die Schwangerschaftsausgänge bei Frauen überwacht, die während der Schwangerschaft SUSTIVA ausgesetzt waren [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Fettumverteilung
Informieren Sie die Patienten darüber, dass es bei Patienten, die eine antiretrovirale Therapie erhalten, zu einer Umverteilung oder Ansammlung von Körperfett kommen kann und dass die Ursache und die langfristigen gesundheitlichen Auswirkungen dieser Erkrankungen nicht bekannt sind [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
SUSTIVA ist eine eingetragene Marke der Bristol-Myers Squibb Pharma Company. ATRIPLA ist eine Marke von Bristol-Myers Squibb & Gilead Sciences, LLC.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Karzinogenese
Mit Efavirenz wurden Langzeitstudien zur Kanzerogenität an Mäusen und Ratten durchgeführt. Mäuse wurden 2 Jahre lang mit 0, 25, 75, 150 oder 300 mg/kg/Tag behandelt. Die Inzidenzen von hepatozellulären Adenomen und Karzinomen sowie pulmonalen alveolären/bronchiolären Adenomen waren bei Frauen über den Normalwert hinaus erhöht. Bei Männern wurde kein Anstieg der Tumorinzidenz über den Hintergrund hinaus beobachtet. Für diese Studie wurde kein NOAEL bei Frauen festgelegt, da bei allen Dosierungen Tumorbefunde auftraten. Die AUC beim NOAEL (150 mg/kg) war bei Männern etwa 0,9-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis. In der Studie an Ratten wurde bei Dosen bis zu 100 mg/kg/Tag, bei denen die AUCs das 0,1- (Männer) oder 0,2-Fache (Frauen) der empfohlenen klinischen Dosis betrugen, kein Anstieg der Tumorinzidenz beobachtet.
Mutagenese
Efavirenz wurde in einer Reihe von In-vitro- und In-vivo-Genotoxizitätstests negativ getestet. Diese umfassten bakterielle Mutationsassays in S. typhimurium und E. coli, Mutationsassays von Säugetieren in Eierstockzellen des chinesischen Hamsters, Chromosomenaberrationsassays in humanen peripheren Blutlymphozyten oder Eierstockzellen des chinesischen Hamsters und ein In-vivo-Maus-Knochenmark-Mikronukleus-Assay.
Beeinträchtigung der Fruchtbarkeit
Efavirenz beeinträchtigte weder die Paarung noch die Fertilität männlicher oder weiblicher Ratten und beeinträchtigte nicht die Spermien behandelter männlicher Ratten. Die Fortpflanzungsfähigkeit von Nachkommen weiblicher Ratten, denen Efavirenz verabreicht wurde, war nicht beeinträchtigt. Die AUCs bei den NOAEL-Werten bei männlichen (200 mg/kg) und weiblichen (100 mg/kg) Ratten waren etwa ≤0,15-mal höher als beim Menschen bei der empfohlenen klinischen Dosis.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Schwangerschafts-Expositionsregister
Es gibt ein Schwangerschafts-Expositionsregister, das den Ausgang der Schwangerschaft bei Frauen überwacht, die während der Schwangerschaft SUSTIVA 600 mg ausgesetzt waren. Ärzte werden ermutigt, Patienten anzumelden, indem sie das Antiretroviral Pregnancy Registry unter 1-800-258-4263 anrufen.
Zusammenfassung der Risiken
Es gibt retrospektive Fallberichte über Neuralrohrdefekte bei Säuglingen, deren Mütter im ersten Trimenon der Schwangerschaft Efavirenz-haltigen Therapien ausgesetzt waren. Prospektive Schwangerschaftsdaten aus dem Antiretroviral Pregnancy Registry reichen nicht aus, um dieses Risiko angemessen einzuschätzen. Verfügbare Daten aus dem Antiretroviral Pregnancy Registry zeigen keinen Unterschied im Gesamtrisiko für schwere Geburtsfehler im Vergleich zur Hintergrundrate für schwere Geburtsfehler von 2,7 % in der US-Referenzpopulation des Metropolitan Atlanta Congenital Defects Program (MACDP). Obwohl kein kausaler Zusammenhang zwischen der Efavirenz-Exposition im ersten Trimenon und Neuralrohrdefekten hergestellt werden konnte, wurden ähnliche Missbildungen in Studien beobachtet, die an Affen mit ähnlichen Dosen wie beim Menschen durchgeführt wurden. Darüber hinaus traten bei Ratten bei einer Dosis, die zehnmal geringer war als die menschliche Exposition bei der empfohlenen klinischen Dosis, fötale und embryonale Toxizitäten auf. Aufgrund des potenziellen Risikos von Neuralrohrdefekten sollte Efavirenz im ersten Trimenon der Schwangerschaft nicht angewendet werden. Informieren Sie schwangere Frauen über das potenzielle Risiko für einen Fötus.
Daten
Menschliche Daten
Es gibt retrospektive Postmarketing-Berichte über Befunde, die mit Neuralrohrdefekten übereinstimmen, einschließlich Meningomyelozele, alle bei Säuglingen von Müttern, die im ersten Trimenon Efavirenz-haltigen Behandlungen ausgesetzt waren.
Basierend auf prospektiven Berichten des Antiretroviral Pregnancy Registry (APR) von etwa 1000 Lebendgeburten nach Exposition gegenüber Efavirenz-haltigen Regimen (einschließlich über 800 Lebendgeburten, die im ersten Trimenon exponiert wurden), gab es keinen Unterschied zwischen Efavirenz und Geburtsfehlern insgesamt im Vergleich zu Efavirenz Hintergrund-Geburtsfehlerrate von 2,7 % in der US-Referenzpopulation des Metropolitan Atlanta Congenital Defects Program. Gemäß dem im Dezember 2014 herausgegebenen Zwischenbericht zum effektiven Jahreszins lag die Prävalenz von Geburtsfehlern nach einer Exposition im ersten Trimester bei 2,3 % (95 %-KI: 1,4 %–3,6 %). Einer dieser prospektiv berichteten Defekte bei Exposition im ersten Trimester war ein Neuralrohrdefekt. Ein einzelner Fall von Anophthalmie mit Efavirenz-Exposition im ersten Trimester wurde ebenfalls prospektiv berichtet. Dieser Fall umfasste auch schwere schräge Gesichtsspalten und Fruchtwasserstreifen, die bekanntermaßen mit Anophthalmie in Verbindung stehen.
Tierdaten
Die Auswirkungen von Efavirenz auf die embryofetale Entwicklung wurden bei drei nichtklinischen Spezies (Cynomolgus-Affen, Ratten und Kaninchen) untersucht. Bei Affen wurde Efavirenz 60 mg/kg/Tag trächtigen Weibchen während der gesamten Trächtigkeit (20. bis 150. Trächtigkeitstag) verabreicht. Die maternale systemische Arzneimittelexposition (AUC) betrug das 1,3-fache der Exposition beim Menschen bei der empfohlenen klinischen Dosis (600 mg/Tag), wobei die fötalen Nabelvenen-Arzneimittelkonzentrationen etwa das 0,7-fache der maternalen Werte betrugen. Drei von 20 Föten/Säuglingen hatten eine oder mehrere Missbildungen; Es gab keine missgebildeten Föten oder Säuglinge von mit Placebo behandelten Müttern. Die Missbildungen, die bei diesen drei Affenföten auftraten, umfassten Anenzephalie und einseitige Anophthalmie bei einem Fötus, Mikrophthalmie bei einem zweiten und eine Gaumenspalte bei dem dritten. Für diese Studie wurde kein NOAEL (No Observable Adverse Effect Level) festgelegt, da nur eine Dosierung bewertet wurde. Bei Ratten wurde Efavirenz entweder während der Organogenese (7. bis 18. Trächtigkeitstag) oder vom 7. Trächtigkeitstag bis zum 21. Laktationstag mit 50, 100 oder 200 mg/kg/Tag verabreicht. Die Verabreichung von 200 mg/kg/Tag an Ratten war mit einem Anstieg der Inzidenz früher Resorptionen verbunden; und Dosen von 100 mg/kg/Tag und mehr waren mit früher neonataler Sterblichkeit verbunden. Die AUC beim NOAEL (50 mg/kg/Tag) war in dieser Rattenstudie 0,1-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis. Die Wirkstoffkonzentrationen in der Milch am 10. Laktationstag waren etwa 8-mal höher als die im mütterlichen Plasma. Bei trächtigen Kaninchen war Efavirenz weder embryoletal noch teratogen, wenn es in Dosierungen von 25, 50 und 75 mg/kg/Tag während der Organogenese (6. bis 18. Trächtigkeitstag) verabreicht wurde. Die AUC beim NOAEL (75 mg/kg/Tag) bei Kaninchen war 0,4-mal so hoch wie beim Menschen bei der empfohlenen klinischen Dosis.
Stillzeit
Zusammenfassung der Risiken
Die Centers for Disease Control and Prevention empfehlen HIV-infizierten Müttern, ihre Säuglinge nicht zu stillen, um eine postnatale Übertragung von HIV zu vermeiden. Wegen der Möglichkeit einer HIV-Übertragung bei gestillten Säuglingen raten Sie Frauen vom Stillen ab.
Weibchen und Männchen mit reproduktivem Potenzial
Wegen möglicher teratogener Wirkungen sollte eine Schwangerschaft bei Frauen, die SUSTIVA erhalten, vermieden werden. [sehen Schwangerschaft .]
Schwangerschaftstest
Frauen im gebärfähigen Alter sollten sich vor Beginn der Behandlung mit SUSTIVA einem Schwangerschaftstest unterziehen.
Empfängnisverhütung
Frauen im gebärfähigen Alter sollten aufgrund der langen Halbwertszeit von Efavirenz während der Behandlung mit SUSTIVA 200 mg und für 12 Wochen nach Absetzen von SUSTIVA 200 mg eine wirksame Verhütungsmethode anwenden. Die Barriere-Kontrazeption sollte immer in Kombination mit anderen Verhütungsmethoden angewendet werden. Hormonelle Methoden, die Progesteron enthalten, können eine verminderte Wirksamkeit haben [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Pädiatrische Verwendung
Die Sicherheit, das pharmakokinetische Profil sowie das virologische und immunologische Ansprechen von SUSTIVA 600 mg wurden bei antiretroviral-naiven und -erfahrenen HIV-1-infizierten pädiatrischen Patienten im Alter von 3 Monaten bis 21 Jahren in drei offenen klinischen Studien untersucht [siehe NEBENWIRKUNGEN , KLINISCHE PHARMAKOLOGIE , und Klinische Studien ]. Die Art und Häufigkeit von Nebenwirkungen in diesen Studien war im Allgemeinen ähnlich wie bei erwachsenen Patienten, mit Ausnahme einer höheren Häufigkeit von Hautausschlag, einschließlich einer höheren Häufigkeit von Hautausschlag Grad 3 oder 4, bei pädiatrischen Patienten im Vergleich zu Erwachsenen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ].
Die Anwendung von SUSTIVA bei Patienten unter 3 Monaten ODER unter 3,5 kg Körpergewicht wird nicht empfohlen, da die Sicherheit, Pharmakokinetik und antivirale Aktivität von SUSTIVA 600 mg in dieser Altersgruppe nicht untersucht wurden und das Risiko besteht, eine HIV-Resistenz zu entwickeln wenn SUSTIVA unterdosiert wird. sehen DOSIERUNG UND ANWENDUNG für Dosierungsempfehlungen für pädiatrische Patienten.
Geriatrische Verwendung
Klinische Studien mit SUSTIVA 200 mg schlossen keine ausreichende Anzahl von Probanden ab 65 Jahren ein, um festzustellen, ob sie anders reagieren als jüngere Probanden. Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein, um die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und von Begleiterkrankungen oder anderen Therapien widerzuspiegeln.
Leberfunktionsstörung
SUSTIVA wird für Patienten mit mäßiger oder schwerer Leberfunktionsstörung nicht empfohlen, da keine ausreichenden Daten vorliegen, um festzustellen, ob eine Dosisanpassung erforderlich ist. Patienten mit leichter Leberfunktionsstörung können ohne Dosisanpassung mit Efavirenz behandelt werden. Aufgrund des umfangreichen Cytochrom-P450-vermittelten Metabolismus von Efavirenz und begrenzter klinischer Erfahrung bei Patienten mit eingeschränkter Leberfunktion ist bei der Verabreichung von SUSTIVA an diese Patienten Vorsicht geboten [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und KLINISCHE PHARMAKOLOGIE ].
ÜBERDOSIS
Einige Patienten, die versehentlich zweimal täglich 600 mg eingenommen haben, haben über verstärkte Symptome des Nervensystems berichtet. Bei einem Patienten traten unwillkürliche Muskelkontraktionen auf.
Die Behandlung einer Überdosierung mit SUSTIVA 200 mg sollte aus allgemeinen unterstützenden Maßnahmen bestehen, einschließlich Überwachung der Vitalfunktionen und Beobachtung des klinischen Zustands des Patienten. Die Verabreichung von Aktivkohle kann verwendet werden, um die Entfernung von nicht resorbiertem Arzneimittel zu unterstützen. Es gibt kein spezifisches Antidot für eine Überdosierung mit SUSTIVA. Da Efavirenz stark an Proteine gebunden ist, ist es unwahrscheinlich, dass eine Dialyse das Arzneimittel signifikant aus dem Blut entfernt.
KONTRAINDIKATIONEN
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Efavirenz ist ein antivirales Medikament [siehe Mikrobiologie ].
Pharmakodynamik
Herzelektrophysiologie
Die Wirkung von SUSTIVA 600 mg auf das QTc-Intervall wurde in einer unverblindeten, positiven und Placebo-kontrollierten Crossover-QT-Studie mit fester Einzelsequenz über 3 Perioden und 3 Behandlungen bei 58 gesunden Probanden, die mit CYP2B6-Polymorphismen angereichert waren, untersucht. Die mittlere Cmax von Efavirenz bei Patienten mit dem CYP2B6 *6/*6-Genotyp nach Verabreichung einer Tagesdosis von 600 mg über 14 Tage war 2,25-mal höher als die mittlere Cmax, die bei Patienten mit dem CYP2B6 *1/*1-Genotyp beobachtet wurde. Es wurde eine positive Beziehung zwischen der Efavirenz-Konzentration und der QTc-Verlängerung beobachtet. Basierend auf der Konzentrations-QTc-Beziehung betragen die mittlere QTc-Verlängerung und ihr oberes 90 %-Konfidenzintervall 8,7 ms und 11,3 ms bei Patienten mit CYP2B6*6/*6-Genotyp nach Verabreichung einer Tagesdosis von 600 mg über 14 Tage [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Pharmakokinetik
Absorption
Maximale Efavirenz-Plasmakonzentrationen von 1,6 – 9,1 μM wurden 5 Stunden nach Verabreichung einer oralen Einzeldosis von 100 mg bis 1600 mg an nicht infizierte Probanden erreicht. Dosisabhängige Anstiege von Cmax und AUC wurden bei Dosen bis zu 1600 mg beobachtet; die Anstiege waren weniger als proportional, was auf eine verringerte Absorption bei höheren Dosen hindeutet.
Bei HIV-1-infizierten Patienten im Steady State waren die mittlere Cmax, mittlere Cmin und mittlere AUC nach Tagesdosen von 200 mg, 400 mg und 600 mg dosisproportional. Die Zeit bis zum Erreichen der maximalen Plasmakonzentrationen betrug etwa 3-5 Stunden und Steady-State-Plasmakonzentrationen wurden in 6-10 Tagen erreicht. Bei 35 Patienten, die SUSTIVA 600 mg einmal täglich erhielten, betrug die Steady-State-Cmax 12,9 ± 3,7 μM (Mittelwert ± Standardabweichung), die Steady-State-Cmin 5,6 ± 3,2 μM und die AUC 184 ± 73 μM•h.
Einfluss der Nahrung auf die orale Aufnahme:
Kapseln
Die Einnahme einer Einzeldosis von 600 mg Efavirenz-Kapseln mit einer fettreichen/kalorienreichen Mahlzeit (894 kcal, 54 g Fett, 54 % Kalorien aus Fett) oder einer fettreduzierten/normalkalorischen Mahlzeit (440 kcal, 2 g Fett, 4 % Kalorien aus Fett) war mit einem mittleren Anstieg der AUC∞ von Efavirenz um 22 % bzw. 17 % und einem mittleren Anstieg der Cmax von Efavirenz um 39 % bzw. 51 % verbunden, relativ zu den Expositionen, die bei Verabreichung im nüchternen Zustand erreicht wurden . [sehen DOSIERUNG UND ANWENDUNG und INFORMATIONEN ZUM PATIENTEN .]
Tablets
Die Einnahme einer einzelnen 600-mg-Efavirenz-Tablette mit einer fettreichen/kalorienreichen Mahlzeit (ca. 1000 kcal, 500-600 kcal aus Fett) war mit einem 28 %igen Anstieg der mittleren AUC∞ von Efavirenz und einem 79 %igen Anstieg des Mittelwertes verbunden Cmax von Efavirenz im Verhältnis zu den unter nüchternen Bedingungen erzielten Expositionen. [sehen DOSIERUNG UND ANWENDUNG und INFORMATIONEN ZUM PATIENTEN .]
Bioverfügbarkeit von Kapselinhalten gemischt mit Nahrungsvehikeln
Bei gesunden erwachsenen Probanden erfüllte die AUC von Efavirenz bei Verabreichung als Inhalt von drei 200-mg-Kapseln gemischt mit 2 Teelöffeln bestimmter Nahrungsvehikel (Apfelmus, Traubengelee oder Joghurt oder Säuglingsnahrung) die Bioäquivalenzkriterien für die AUC der verabreichten intakten Kapselformulierung unter nüchternen Bedingungen.
Verteilung
Efavirenz wird in hohem Maße (ca. 99,5-99,75 %) an menschliche Plasmaproteine, überwiegend Albumin, gebunden. Bei HIV-1-infizierten Patienten (n = 9), die SUSTIVA 200 bis 600 mg einmal täglich für mindestens einen Monat erhielten, lagen die Konzentrationen im Liquor cerebrospinalis im Bereich von 0,26 bis 1,19 % (Mittelwert 0,69 %) der entsprechenden Plasmakonzentration. Dieser Anteil ist etwa 3-mal höher als der nicht proteingebundene (freie) Anteil von Efavirenz im Plasma.
Stoffwechsel
Studien am Menschen und In-vitro-Studien mit menschlichen Lebermikrosomen haben gezeigt, dass Efavirenz hauptsächlich durch das Cytochrom-P450-System zu hydroxylierten Metaboliten mit anschließender Glucuronidierung dieser hydroxylierten Metaboliten metabolisiert wird. Diese Metaboliten sind gegenüber HIV-1 im Wesentlichen inaktiv. Die In-vitro-Studien deuten darauf hin, dass CYP3A und CYP2B6 die wichtigsten Isozyme sind, die für den Metabolismus von Efavirenz verantwortlich sind.
Es wurde gezeigt, dass Efavirenz CYP-Enzyme induziert, was zu einer Induktion seines eigenen Metabolismus führt. Mehrfachdosen von 200–400 mg pro Tag über 10 Tage führten zu einem geringeren Ausmaß der Akkumulation als erwartet (22–42 % niedriger) und einer kürzeren terminalen Halbwertszeit von 40–55 Stunden (Halbwertszeit einer Einzeldosis 52–76 Stunden). ).
Beseitigung
Efavirenz hat eine terminale Halbwertszeit von 52-76 Stunden nach Einzeldosen und 40-55 Stunden nach Mehrfachdosen. Es wurde eine einmonatige Massenbilanz/Ausscheidungsstudie mit 400 mg pro Tag mit einer C-markierten Dosis durchgeführt, die an Tag 8 verabreicht wurde. Ungefähr 14-34 % der radioaktiv markierten Substanz wurden im Urin und 16-61 % im Stuhl wiedergefunden . Fast die gesamte Urinausscheidung des radioaktiv markierten Arzneimittels erfolgte in Form von Metaboliten. Efavirenz machte den größten Teil der gesamten im Stuhl gemessenen Radioaktivität aus.
Besondere Populationen
Pädiatrie
Die pharmakokinetischen Parameter für Efavirenz im Steady State bei pädiatrischen Patienten wurden durch ein populationspharmakokinetisches Modell vorhergesagt und sind in Tabelle 6 nach Gewichtsbereichen zusammengefasst, die den empfohlenen Dosen entsprechen.
Geschlecht und Rasse
Die Pharmakokinetik von Efavirenz bei Patienten scheint bei Männern und Frauen sowie bei den untersuchten ethnischen Gruppen ähnlich zu sein.
Nierenfunktionsstörung
Die Pharmakokinetik von Efavirenz wurde bei Patienten mit Niereninsuffizienz nicht untersucht; jedoch wird weniger als 1 % von Efavirenz unverändert im Urin ausgeschieden, sodass die Auswirkungen einer Nierenfunktionsstörung auf die Efavirenz-Elimination minimal sein sollten.
Leberfunktionsstörung
Eine Mehrfachdosisstudie zeigte keine signifikante Wirkung auf die Pharmakokinetik von Efavirenz bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) im Vergleich zu Kontrollen. Es lagen keine ausreichenden Daten vor, um festzustellen, ob eine mäßige oder schwere Leberfunktionsstörung (Child-Pugh-Klasse B oder C) die Pharmakokinetik von Efavirenz beeinflusst.
Arzneimittelwechselwirkungsstudien
Es wurde in vivo gezeigt, dass Efavirenz eine hepatische Enzyminduktion verursacht und somit die Biotransformation einiger Arzneimittel erhöht, die durch CYP3A und CYP2B6 metabolisiert werden. In-vitro-Studien haben gezeigt, dass Efavirenz die CYP-Isoenzyme 2C9 und 2C19 mit Ki-Werten (8,5-17 μM) im Bereich der beobachteten Efavirenz-Plasmakonzentrationen hemmt. In In-vitro-Studien hemmte Efavirenz CYP2E1 nicht und hemmte CYP2D6 und CYP1A2 (Ki-Werte 82-160 μM) nur bei Konzentrationen, die deutlich über den klinisch erreichten liegen. Die gleichzeitige Anwendung von Efavirenz mit Arzneimitteln, die hauptsächlich durch CYP2C9-, CYP2C19-, CYP3A- oder CYP2B6-Isozyme metabolisiert werden, kann zu veränderten Plasmakonzentrationen des gleichzeitig verabreichten Arzneimittels führen. Es ist zu erwarten, dass Arzneimittel, die CYP3A- und CYP2B6-Aktivität induzieren, die Clearance von Efavirenz erhöhen, was zu verringerten Plasmakonzentrationen führt.
Wechselwirkungsstudien wurden mit Efavirenz und anderen Arzneimitteln durchgeführt, die wahrscheinlich gleichzeitig verabreicht werden, oder Arzneimitteln, die üblicherweise als Sonden für pharmakokinetische Wechselwirkungen verwendet werden. Die Wirkungen der gleichzeitigen Verabreichung von Efavirenz auf Cmax, AUC und Cmin sind in Tabelle 7 (Wirkung von Efavirenz auf andere Arzneimittel) und Tabelle 8 (Wirkung anderer Arzneimittel auf Efavirenz) zusammengefasst. Informationen zu klinischen Empfehlungen finden Sie unter WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN .
Mikrobiologie
Wirkmechanismus
Efavirenz ist ein NNRTI von HIV-1. Die Aktivität von Efavirenz wird überwiegend durch nicht-kompetitive Hemmung der Reversen Transkriptase von HIV-1 vermittelt. Die reverse Transkriptase von HIV-2 und die humanen zellulären DNA-Polymerasen α, β, γ und δ werden durch Efavirenz nicht gehemmt.
Antivirale Aktivität in der Zellkultur
Die Konzentration von Efavirenz, die die Replikation von laboradaptierten Wildtypstämmen und klinischen Isolaten in Zellkulturen um 90-95 % (EC90-95) hemmt, lag im Bereich von 1,7 bis 25 nM in lymphoblastoiden Zelllinien, peripheren mononukleären Blutzellen (PBMCs) und Makrophagen /Monozytenkulturen. Efavirenz zeigte eine antivirale Aktivität gegen Klade B- und die meisten Nicht-Klade B-Isolate (Subtypen A, AE, AG, C, D, F, G, J, N), hatte aber eine reduzierte antivirale Aktivität gegen Gruppe-O-Viren. Efavirenz zeigte in Zellkultur eine additive antivirale Aktivität ohne Zytotoxizität gegen HIV-1, wenn es mit den NNRTIs Delavirdin und Nevirapin, NRTIs (Abacavir, Didanosin, Emtricitabin, Lamivudin, Stavudin, Tenofovir, Zalcitabin, Zidovudin), PIs (Amprenavir, Indinavir, Lopinavir, Nelfinavir, Ritonavir, Saquinavir) und der Fusionsinhibitor Enfuvirtid. Efavirenz zeigte in Zellkultur mit Atazanavir eine additive bis antagonistische antivirale Aktivität. Efavirenz war nicht antagonistisch mit Adefovir, das zur Behandlung einer Hepatitis-B-Virusinfektion angewendet wird, oder mit Ribavirin, das in Kombination mit Interferon zur Behandlung einer Hepatitis-C-Virusinfektion angewendet wird.
Widerstand
In Zellkultur
In Zellkulturen traten in Gegenwart des Arzneimittels schnell HIV-1-Isolate mit reduzierter Empfindlichkeit gegenüber Efavirenz (>380-facher Anstieg des EC90-Werts) auf. Die genotypische Charakterisierung dieser Viren identifizierte einzelne Aminosäuresubstitutionen L100I oder V179D, doppelte Substitutionen L100I/V108I und dreifache Substitutionen L100I/V179D/Y181C in der reversen Transkriptase.
Klinische Studien
Klinische Isolate mit reduzierter Empfindlichkeit gegenüber Efavirenz in Zellkulturen wurden erhalten. Eine oder mehrere Substitutionen an den Aminosäurepositionen 98, 100, 101, 103, 106, 108, 188, 190, 225 und 227 in der Reversen Transkriptase wurden bei Patienten beobachtet, bei denen die Behandlung mit Efavirenz in Kombination mit Indinavir oder mit Zidovudin plus Lamivudin fehlschlug. Die K103N-Substitution wurde am häufigsten beobachtet. In der langfristigen Resistenzüberwachung (durchschnittlich 52 Wochen, Bereich 4–106 Wochen) wurden 28 Isolate mit übereinstimmenden Ausgangswerten und virologischem Versagen analysiert. Einundsechzig Prozent (17/28) dieser fehlgeschlagenen Isolate hatten eine verringerte Efavirenz-Empfindlichkeit in der Zellkultur mit einer medianen 88-fachen Veränderung der Efavirenz-Empfindlichkeit (EC50-Wert) gegenüber der Referenz. Die häufigste NNRTI-Substitution, die sich bei diesen Patientenisolaten entwickelte, war K103N (54 %). Andere NNRTI-Substitutionen, die sich entwickelten, waren L100I (7 %), K101E/Q/R (14 %), V108I (11 %), G190S/T/A (7 %), P225H (18 %) und M230I/L (11 %).
Kreuzresistenz
Kreuzresistenz zwischen NNRTIs wurde beobachtet. Klinische Isolate, die zuvor als Efavirenz-resistent charakterisiert wurden, waren im Vergleich zum Ausgangswert in Zellkulturen auch phänotypisch resistent gegen Delavirdin und Nevirapin. Delavirdin- und/oder Nevirapin-resistente klinische Virusisolate mit NNRTI-Resistenz-assoziierten Substitutionen (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L oder M230L) zeigten eine reduzierte Empfindlichkeit gegenüber Efavirenz in Zellkultur. Mehr als 90 % der in Zellkultur getesteten NRTI-resistenten klinischen Isolate behielten ihre Empfindlichkeit gegenüber Efavirenz.
Tiertoxikologie
Nicht anhaltende Krämpfe wurden bei 6 von 20 Affen beobachtet, die Efavirenz in Dosen erhielten, die Plasma-AUC-Werte ergaben, die 4- bis 13-mal höher waren als beim Menschen, der die empfohlene Dosis erhielt [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Klinische Studien
Erwachsene
Studie 006 eine randomisierte, offene Studie, verglich SUSTIVA (600 mg einmal täglich) + Zidovudin (ZDV, 300 mg alle 12 Stunden) + Lamivudin (LAM, 150 mg alle 12 Stunden) oder SUSTIVA (600 mg einmal täglich) + Indinavir (IDV, 1000 mg q8h) mit Indinavir (800 mg q8h) + Zidovudin (300 mg q12h) + Lamivudin (150 mg q12h). Zwölfhundertsechsundsechzig Patienten (Durchschnittsalter 36,5 Jahre [Bereich 18–81], 60 % Kaukasier, 83 % männlich) wurden eingeschlossen. Alle Patienten waren bei Studieneintritt Efavirenz-, Lamivudin-, NNRTI- und PI-naiv. Die mediane CD4+-Zellzahl zu Studienbeginn betrug 320 Zellen/mm3 und die mediane HIV-1-RNA-Konzentration zu Studienbeginn betrug 4,8 log Kopien/ml. Die Behandlungsergebnisse mit dem Standard-Assay (Assay-Grenze 400 Kopien/ml) über 48 und 168 Wochen sind in Tabelle 9 dargestellt. Versionen des AMPLICOR HIV-1 MONITOR Assays. Während der Studie wurde Version 1.5 des Assays in Europa eingeführt, um den Nachweis von Nicht-Clade-B-Viren zu verbessern.
Bei Patienten, die mit SUSTIVA + Zidovudin + Lamivudin, SUSTIVA + Indinavir oder Indinavir + Zidovudin + Lamivudin behandelt wurden, betrug der Prozentsatz der Responder mit HIV-1-RNA
ACTG 364 ist eine randomisierte, doppelblinde, placebokontrollierte 48-wöchige Studie an NRTI-erfahrenen Patienten, die zwei vorherige ACTG-Studien abgeschlossen hatten. Einhundertsechsundneunzig Patienten (Durchschnittsalter 41 Jahre [Bereich 18–76], 74 % Kaukasier, 88 % männlich) erhielten NRTIs in Kombination mit SUSTIVA (600 mg einmal täglich) oder Nelfinavir (NFV, 750 mg dreimal täglich). ) oder SUSTIVA (600 mg einmal täglich) + Nelfinavir randomisiert und doppelblind verabreicht. Die mittlere CD4+-Zellzahl zu Studienbeginn betrug 389 Zellen/mm3 und die mittlere HIV-1-RNA-Konzentration zu Studienbeginn 8130 Kopien/ml. Bei Eintritt in die Studie wurde allen Patienten ein neues offenes NRTI-Regime zugewiesen, das von ihrer bisherigen NRTI-Behandlungserfahrung abhängig war. Es gab keinen signifikanten Unterschied in der mittleren CD4+-Zellzahl zwischen den Behandlungsgruppen; die durchschnittliche Gesamtzunahme betrug etwa 100 Zellen nach 48 Wochen bei Patienten, die die Studienbehandlungen fortsetzten. Die Behandlungsergebnisse sind in Tabelle 10 aufgeführt. Die Plasma-HIV-RNA-Spiegel wurden mit dem AMPLICOR HIV-1 MONITOR-Assay unter Verwendung einer unteren Quantifizierungsgrenze von 500 Kopien/ml quantifiziert.
Eine Kaplan-Meier-Analyse der Zeit bis zum Therapieversagen über 72 Wochen zeigt eine längere Dauer der virologischen Suppression (HIV-RNA
Pädiatrische Patienten
Studie AI266922 ist eine Open-Label-Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA 200 mg in Kombination mit Didanosin und Emtricitabin bei antiretroviral-naiven und -erfahrenen pädiatrischen Patienten. 37 Patienten im Alter von 3 Monaten bis 6 Jahren (Median 0,7 Jahre) wurden mit SUSTIVA behandelt. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 5,88 log Kopien/ml, die mediane CD4+-Zellzahl 1144 Zellen/mm3 und der mediane CD4+-Prozentsatz 25 %. Die mediane Dauer der Studientherapie betrug 60 Wochen; 27 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrugen die Gesamtanteile von Patienten mit HIV-RNA
Studie PACTG 1021 war eine offene Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA 200 mg in Kombination mit Didanosin und Emtricitabin bei pädiatrischen Patienten, die keine antiretrovirale Therapie erhalten hatten. 43 Patienten im Alter von 3 Monaten bis 21 Jahren (Median 9,6 Jahre) erhielten eine Dosis von SUSTIVA. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 4,8 log Kopien/ml, die mediane CD4+-Zellzahl 367 Zellen/mm3 und der mediane CD4+-Prozentsatz 18 %. Die mediane Dauer der Studientherapie betrug 181 Wochen; 16 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrugen die Gesamtanteile von Patienten mit HIV-RNA
Studie PACTG 382 war eine offene Studie zur Bewertung der Pharmakokinetik, Sicherheit, Verträglichkeit und antiviralen Aktivität von SUSTIVA in Kombination mit Nelfinavir und einem NRTI bei antiretroviral-naiven und NRTI-erfahrenen pädiatrischen Patienten. 102 Patienten im Alter von 3 Monaten bis 16 Jahren (Median 5,7 Jahre) wurden mit SUSTIVA behandelt. 87 % der Patienten hatten zuvor eine antiretrovirale Therapie erhalten. Zu Studienbeginn betrug die mediane Plasma-HIV-1-RNA 4,57 log Kopien/ml, die mediane CD4+-Zellzahl 755 Zellen/mm3 und der mediane CD4+-Prozentsatz 30 %. Die mediane Dauer der Studientherapie betrug 118 Wochen; 25 % der Patienten brachen die Behandlung vor Woche 48 ab. Gemäß einer ITT-Analyse betrug der Gesamtanteil der Patienten mit HIV-RNA
INFORMATIONEN ZUM PATIENTEN
SUSTIVA® (sus-TEE-vah) (Efavirenz) Kapseln
SUSTIVA® (sus-TEE-vah) (Efavirenz) Tabletten
Wichtig: Fragen Sie Ihren Arzt oder Apotheker nach Arzneimitteln, die Sie nicht zusammen mit SUSTIVA einnehmen sollten. Weitere Informationen finden Sie im Abschnitt „Was soll ich meinem Arzt vor der Einnahme von SUSTIVA mitteilen?“
Lesen Sie diese Patienteninformation, bevor Sie mit der Einnahme von SUSTIVA beginnen und jedes Mal, wenn Sie eine Nachfüllung erhalten. Möglicherweise gibt es neue Informationen. Diese Informationen ersetzen nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung.
Was ist SUSTIVA?
SUSTIVA 600 mg ist ein verschreibungspflichtiges HIV-1-Arzneimittel (Humanes Immunschwächevirus Typ 1), das zusammen mit anderen antiretroviralen Arzneimitteln zur Behandlung einer HIV-1-Infektion bei Erwachsenen und Kindern angewendet wird, die mindestens 3 Monate alt sind und mindestens 7 Pfund 12 Unzen (3,5 kg) wiegen kg). HIV ist das Virus, das AIDS (Acquired Immune Deficiency Syndrome) verursacht.
Es ist nicht bekannt, ob SUSTIVA bei Kindern unter 3 Monaten oder mit einem Gewicht von weniger als 3,5 kg (7 Pfund 12 Unzen) sicher und wirksam ist.
Bei Anwendung mit anderen antiretroviralen Arzneimitteln zur Behandlung einer HIV-1-Infektion kann SUSTIVA 600 mg helfen:
Die Reduzierung der HIV-1-Menge und die Erhöhung der CD4+ (T)-Zellen in Ihrem Blut kann zur Verbesserung Ihres Immunsystems beitragen. Dies kann Ihr Risiko zu sterben oder Infektionen zu bekommen, die auftreten können, wenn Ihr Immunsystem geschwächt ist (opportunistische Infektionen).
SUSTIVA 600 mg heilt weder eine HIV-1-Infektion noch AIDS. Sie sollten weiterhin HIV-1-Medikamente einnehmen, um eine HIV-1-Infektion zu kontrollieren und HIV-bedingte Erkrankungen zu verringern.
Vermeiden Sie Dinge, die eine HIV-1-Infektion auf andere übertragen können:
Fragen Sie Ihren Arzt, wenn Sie Fragen dazu haben, wie Sie verhindern können, dass HIV auf andere Menschen übertragen wird.
Wer sollte SUSTIVA 600 mg nicht einnehmen?
Nehmen Sie SUSTIVA nicht ein, wenn Sie allergisch gegen Efavirenz oder einen der sonstigen Bestandteile von SUSTIVA sind. Eine vollständige Liste der Inhaltsstoffe von SUSTIVA finden Sie am Ende dieser Packungsbeilage.
Nehmen Sie SUSTIVA nicht ein, wenn Sie derzeit Elbasvir und Grazoprevir (ZEPATIER®) einnehmen.
Was sollte ich meinem Arzt vor der Einnahme von SUSTIVA 200 mg mitteilen?
Informieren Sie vor der Einnahme von SUSTIVA Ihren Arzt, wenn Sie an einer Erkrankung leiden und insbesondere, wenn Sie:
Informieren Sie Ihren Arzt und Apotheker über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und Kräuterergänzungen.
SUSTIVA 600 mg kann die Wirkungsweise anderer Arzneimittel beeinflussen, und andere Arzneimittel können die Wirkungsweise von SUSTIVA 600 mg beeinflussen. und kann schwerwiegende Nebenwirkungen verursachen. Wenn Sie bestimmte Arzneimittel zusammen mit SUSTIVA einnehmen, kann die Menge an SUSTIVA 600 mg in Ihrem Körper zu niedrig sein und es kann möglicherweise nicht dazu beitragen, Ihre HIV-Infektion zu kontrollieren. Das HIV-Virus in Ihrem Körper kann gegen SUSTIVA oder andere ähnliche HIV-Arzneimittel resistent werden.
Sie sollten SUSTIVA 600 mg nicht einnehmen, wenn Sie ATRIPLA einnehmen (Efavirenz, Emtricitabin, Tenofovirdisoproxilfumarat), es sei denn, Ihr Arzt hat es Ihnen gesagt.
Informieren Sie Ihren Arzt und Apotheker über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und Kräuterergänzungen. Einige Arzneimittel interagieren mit SUSTIVA.
Führen Sie eine Liste Ihrer Arzneimittel, um sie Ihrem Arzt und Apotheker zu zeigen.
Wie soll ich SUSTIVA einnehmen?
Wie und wann ist SUSTIVA einzunehmen?
Welche Nebenwirkungen kann SUSTIVA haben?
SUSTIVA 600 mg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Wenn Sie unter Schwindel, Konzentrationsschwierigkeiten oder Benommenheit leiden, fahren Sie kein Auto, bedienen Sie keine Maschinen und tun Sie nichts, bei dem Sie wachsam sein müssen.
Einige Symptome des Nervensystems (z. B. Verwirrtheit, verlangsamte Gedanken und körperliche Bewegungen sowie Wahnvorstellungen [falsche Überzeugungen] oder Halluzinationen [Sehen oder Hören von Dingen, die andere nicht sehen oder hören]) können Monate bis Jahre nach Beginn der SUSTIVA 600 mg-Therapie auftreten. Wenden Sie sich umgehend an Ihren Arzt, wenn eines dieser Symptome auftritt.
Informieren Sie sofort Ihren Arzt, wenn bei Ihnen eines der folgenden Symptome auftritt:
Zu den häufigsten Nebenwirkungen von SUSTIVA 600 mg gehören:
Bei einigen Patienten, die SUSTIVA einnahmen, traten erhöhte Blutfettwerte (Cholesterin und Triglyceride) auf. Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von SUSTIVA. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Wie ist SUSTIVA aufzubewahren?
Bewahren Sie SUSTIVA und alle Arzneimittel für Kinder unzugänglich auf.
Allgemeine Informationen zu SUSTIVA
Arzneimittel werden manchmal zu anderen als den in der Packungsbeilage aufgeführten Zwecken verschrieben. Verwenden Sie SUSTIVA 600 mg nicht für eine Erkrankung, für die es nicht verschrieben wurde. Geben Sie SUSTIVA 600 mg nicht an andere Personen, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Wenn Sie weitere Informationen wünschen, sprechen Sie mit Ihrem Arzt. Sie können Ihren Apotheker oder Arzt um Informationen über SUSTIVA bitten, die für Angehörige der Gesundheitsberufe bestimmt sind. Weitere Informationen erhalten Sie unter www.sustiva.com oder telefonisch unter 1-800-321-1335.
Welche Inhaltsstoffe enthält SUSTIVA?
Wirkstoff: Efavirenz
Inaktive Zutaten:
SUSTIVA 600 mg Kapseln: Lactosemonohydrat, Magnesiumstearat, Natriumlaurylsulfat und Natriumstärkeglykolat. Die Kapselhülle enthält Gelatine, Natriumlaurylsulfat, Titandioxid und/oder gelbes Eisenoxid. Die Kapselhülle kann auch Siliziumdioxid enthalten. Die Kapseln sind mit Tinte bedruckt, die Carmine 40 Blue, FD&C Blue No. 2 und Titandioxid enthält.
SUSTIVA-Tabletten: Croscarmellose-Natrium, Hydroxypropylcellulose, Lactosemonohydrat, Magnesiumstearat, mikrokristalline Cellulose und Natriumlaurylsulfat. Die Filmbeschichtung der Tablette enthält Opadry Yellow und Opadry Clear. Die Tabletten sind mit Carnaubawachs poliert und mit violetter Tinte, Opacode WB, bedruckt.
Diese Patienteninformation wurde von der US Food and Drug Administration genehmigt.
Gebrauchsanweisung
SUSTIVA® (sus-TEE-vah) (Efavirenz) Kapseln
Zubereitung einer Dosis von SUSTIVA 600 mg nach der Kapselstreumethode
Lesen Sie diese Gebrauchsanweisung, bevor Sie Ihre erste Dosis SUSTIVA gemischt mit Nahrung oder Säuglingsanfangsnahrung nach der Kapselstreumethode zubereiten, jedes Mal, wenn Sie eine Nachfüllung erhalten, und bei Bedarf. Möglicherweise gibt es neue Informationen. Diese Informationen ersetzen nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung. Wenden Sie sich an Ihren Arzt oder Apotheker, wenn Sie Fragen zum Mischen oder Verabreichen einer Dosis von SUSTIVA 200 mg nach der Kapselsprühmethode haben.
Wichtige Informationen:
Zubereitung einer Dosis von SUSTIVA 600 mg gemischt mit Nahrung nach der Kapselstreumethode.
Bevor Sie eine Dosis von SUSTIVA 200 mg gemischt mit Nahrung nach der Kapselstreumethode zubereiten, besorgen Sie die folgenden Vorräte:
Schritt 1. Wählen Sie eine saubere, ebene Arbeitsfläche. Legen Sie ein sauberes Papiertuch auf die Arbeitsfläche. Legen Sie dann die anderen Vorräte auf das Papiertuch.
Schritt 2. Waschen und trocknen Sie Ihre Hände gut.
Schritt 3. Geben Sie 1 bis 2 Teelöffel weiche Lebensmittel wie Apfelmus, Traubengelee oder Joghurt in den kleinen Behälter (siehe Abbildung A ). Die Farbe und Dicke des Lebensmittels kann sich ändern, wenn es mit dem Arzneimittel gemischt wird.
Abbildung A
Schritt 4. Es gibt 2 Teile der SUSTIVA 200 mg Kapsel. Sehen Sie sich die SUSTIVA-Kapsel an, um zu sehen, welcher Teil der Kapsel den anderen Teil überlappt (siehe Abbildung B ).
Abbildung B
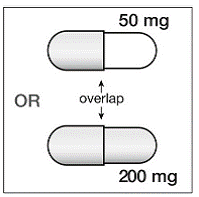
Schritt 5. Halten Sie die SUSTIVA-Kapsel seitlich (waagerecht) direkt über den Lebensmittelbehälter. Halten Sie jedes Ende der SUSTIVA-Kapsel zwischen Daumen und Zeigefinger (siehe Abbildung C ).
Abbildung C
Schritt 6. Verwenden Sie Ihren Daumen und Zeigefinger, um nahe dem Ende des überlappenden Teils der SUSTIVA 600-mg-Kapsel zusammenzudrücken (siehe Abbildung D ).
Abbildung D
Drehen Sie dann beide Enden der SUSTIVA 600 mg-Kapsel vorsichtig in entgegengesetzte Richtungen, um sie zu öffnen (siehe Abbildung E ). Achten Sie darauf, den Kapselinhalt nicht zu verschütten oder in der Luft zu verteilen.
Abbildung E

Schritt 7. Streuen Sie den Inhalt der SUSTIVA-Kapsel auf das Futter (siehe Abbildung F ).
Abbildung F

Wenn die verschriebene Gesamtdosis mehr als 1 Kapsel beträgt, befolgen Sie die Schritte 4 bis 7 für jede Kapsel. Fügen Sie keine weiteren Lebensmittel hinzu.
Die Schritte 8 bis 11 sollten abgeschlossen sein innerhalb von 30 Minuten des Mischens der Medizin (siehe Abbildung G ).
Abbildung G

Schritt 8. Verwenden Sie den kleinen Löffel, um den Kapselinhalt und die Nahrung vorsichtig miteinander zu vermischen (siehe Abbildung H ). Streusel lösen sich nicht auf. Die Mischung sieht körnig aus, sollte aber nicht klumpig sein.
Abbildung H
Schritt 9. Verwenden Sie den kleinen Löffel, um die Mischung aus Nahrung und Kapselinhalt zu verabreichen oder einzunehmen. Stellen Sie sicher, dass die gesamte Mischung geschluckt wird.
Schritt 10. Geben Sie weitere 2 Teelöffel der Nahrung in den leeren Behälter und rühren Sie vorsichtig mit dem kleinen Löffel um, um sich mit dem eventuell noch im Behälter befindlichen Kapselinhalt zu vermischen.
Schritt 11. Verwenden Sie den kleinen Löffel, um die Mischung aus Nahrung und Kapselinhalt zu verabreichen oder einzunehmen. Stellen Sie sicher, dass die gesamte Mischung geschluckt wird.
Schritt 12. Behälter und Löffel waschen. Werfen Sie das Papiertuch weg und reinigen Sie die Arbeitsfläche. Wasche deine Hände.
Zubereitung einer Dosis von SUSTIVA 600 mg gemischt mit Säuglingsanfangsnahrung nach der Kapselstreumethode
Um sicherzustellen, dass Ihr Baby das gesamte Arzneimittel erhält, geben Sie Ihrem Baby den Inhalt der SUSTIVA-Kapsel nicht in einer Flasche.
Bevor Sie eine Dosis SUSTIVA gemischt mit Säuglingsanfangsnahrung nach der Kapselstreumethode zubereiten, besorgen Sie die folgenden Vorräte:
Abbildung I
Schritt 1. Bereiten Sie die Säuglingsnahrung gemäß den Anweisungen auf der Packung der Säuglingsnahrung zu. Sie werden etwa 1 Unze der Formel verwenden, um das Arzneimittel zu verabreichen. Restliche Formel sollte dem Kind 2 Stunden lang nicht gegeben werden.
Schritt 2. Wählen Sie eine saubere, ebene Arbeitsfläche. Legen Sie ein sauberes Papiertuch auf die Arbeitsfläche. Legen Sie die Materialien, die Sie benötigen, auf das Papiertuch.
Schritt 3. Waschen und trocknen Sie Ihre Hände gut.
Schritt 4. Gießen Sie 2 Teelöffel zimmerwarme Säuglingsnahrung in den Behälter (siehe Abbildung J ).
Abbildung J
Schritt 5. Die SUSTIVA-Kapsel besteht aus 2 Teilen. Sehen Sie sich die SUSTIVA-Kapsel an, um zu sehen, welcher Teil der Kapsel den anderen Teil überlappt (siehe Abbildung K ).
Abbildung K
Schritt 6. Halten Sie die SUSTIVA-Kapsel seitlich (waagerecht) direkt über das Behältnis mit der Säuglingsanfangsnahrung. Halten Sie jedes Ende der SUSTIVA 600 mg-Kapsel zwischen Daumen und Zeigefinger (siehe Abbildung L ).
Abbildung L
Schritt 7. Drücken Sie mit Daumen und Zeigefinger nahe dem Ende des überlappenden Teils der SUSTIVA-Kapsel zusammen (siehe Abbildung M ).
Abbildung M
Drehen Sie dann beide Enden der SUSTIVA-Kapsel vorsichtig in entgegengesetzte Richtungen, um sie zu öffnen (siehe Abbildung N ). Achten Sie darauf, den Kapselinhalt nicht zu verschütten oder in der Luft zu verteilen.
Abbildung N
Schritt 8. Streuen Sie den Inhalt der SUSTIVA 600 mg Kapsel auf die Säuglingsnahrung (siehe Abbildung O ).
Abbildung O
Wenn die verschriebene Gesamtdosis mehr als 1 Kapsel beträgt, befolgen Sie die Schritte 5 bis 8 für jede Kapsel. Fügen Sie keine weitere Säuglingsnahrung hinzu.
Die Schritte 9 bis 12 sollten abgeschlossen sein innerhalb von 30 Minuten des Mischens der Medizin (siehe Abbildung P ).
Abbildung P

Schritt 9. Halten Sie den Behälter mit einer Hand. Verwenden Sie mit der anderen Hand den Teelöffel, um den Kapselinhalt und die Säuglingsnahrung vorsichtig zu vermischen (siehe Abbildung Q ). Streusel lösen sich nicht auf. Die Mischung sieht körnig aus, sollte aber nicht klumpig sein.
Abbildung Q
Schritt 10. So ziehen Sie die gesamte Mischung in die Applikationsspritze für Zubereitungen zum Einnehmen auf:
Abbildung R
Abbildung S
Abbildung T
Schritt 11. Setzen Sie die Spitze der Spritze in den Mund Ihres Babys entlang der inneren Wange (siehe Abbildung u ). Drücken Sie langsam auf den Kolben, um Ihrem Baby die gesamte Mischung zu geben.
Abbildung u
Schritt 12. Um sicherzustellen, dass Ihr Baby das gesamte Arzneimittel erhält:
Abbildung V
Schritt 13. Entfernen Sie den Kolben von der Applikationsspritze für Zubereitungen zum Einnehmen. Waschen Sie das Behältnis, den Teelöffel und die orale Dosierspritze. Lassen Sie den Kolben und den Spritzenzylinder trocknen, bevor Sie sie wieder zusammensetzen.
Schritt 14. Werfen Sie das Papiertuch weg und reinigen Sie die Arbeitsfläche. Wasche deine Hände.
Wie soll ich SUSTIVA 600 mg Kapseln aufbewahren?
Bewahren Sie SUSTIVA 600 mg Kapseln und alle Arzneimittel außerhalb der Reichweite von Kindern auf.
Diese Gebrauchsanweisung wurde von der US Food and Drug Administration genehmigt.
Was ist Symbicort 4,5 mcg und wie wird es angewendet?
Symbicort 4,5 mcg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Asthma und chronisch obstruktiver Lungenerkrankung. Symbicort kann allein oder mit anderen Medikamenten verwendet werden.
Symbicort gehört zu einer Klasse von Medikamenten, die Respiratory Inhalant Combos genannt werden.
Es ist nicht bekannt, ob Symbicort 4,5 µg bei Kindern unter 6 Jahren sicher und wirksam ist.
Welche Nebenwirkungen kann Symbicort 4,5 mcg haben?
Symbicort kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Symbicort gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Symbicort. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
SYMBICORT 80/4.5 und SYMBICORT 160/4.5 enthalten jeweils mikronisiertes Budesonid und mikronisiertes Formoterolfumarat-Dihydrat nur zur oralen Inhalation.
Jeder SYMBICORT 80/4.5- und SYMBICORT 160/4.5-Kanister ist als ein mit Hydrofluoralkan (HFA 227; 1,1,1,2,3,3,3-Heptafluorpropan) angetriebener, unter Druck stehender Dosierinhalator formuliert, der entweder 60 oder 120 Sprühstöße enthält [siehe Darreichungsformen und Stärken und WIE GELIEFERT / Lagerung und Handhabung ]. Nach dem Vorfüllen dosiert jede Betätigung entweder 91/5,1 µg oder 181/5,1 µg vom Ventil und gibt entweder 80/4,5 µg oder 160/4,5 µg (mikronisiertes Budesonid/mikronisiertes Formoterolfumarat-Dihydrat) vom Auslöser ab. Die tatsächliche an die Lunge abgegebene Arzneimittelmenge kann von Faktoren des Patienten abhängen, wie z. B. der Koordination zwischen der Betätigung der Vorrichtung und dem Einatmen durch das Abgabesystem. SYMBICORT 160 mg enthält außerdem Povidon K25 USP als Suspendiermittel und Polyethylenglykol 1000 NF als Gleitmittel.
SYMBICORT 160 mg sollte vor der ersten Anwendung vorbereitet werden, indem zwei Testsprühstöße vom Gesicht weg in die Luft abgegeben werden und vor jedem Sprühstoß 5 Sekunden lang gut geschüttelt wird. In Fällen, in denen der Inhalator länger als 7 Tage nicht verwendet wurde oder wenn er fallen gelassen wurde, bereiten Sie den Inhalator erneut vor, indem Sie ihn vor jedem Sprühstoß 5 Sekunden lang gut schütteln und zwei Testsprühstöße vom Gesicht weg in die Luft abgeben.
Ein Wirkstoff von SYMBICORT 4,5 µg ist Budesonid, ein Kortikosteroid, das chemisch als (RS)11β, 16α, 17,21-Tetrahydroxypregna-1,4-dien-3,20-dion zyklisches 16,17-acetal mit Butyraldehyd bezeichnet wird. Budesonid wird als Mischung aus zwei Epimeren (22R und 22S) bereitgestellt. Die Summenformel von Budesonid ist C25H34O6 und sein Molekulargewicht beträgt 430,5. Seine Strukturformel lautet:
Budesonid ist ein weißes bis cremefarbenes, geschmacks- und geruchloses Pulver, das in Wasser und Heptan praktisch unlöslich, in Ethanol schwer löslich und in Chloroform gut löslich ist. Sein Verteilungskoeffizient zwischen Octanol und Wasser bei pH 7,4 beträgt 1,6 x 103.
Der andere Wirkstoff von SYMBICORT 160 mg ist Formoterolfumarat-Dihydrat, ein selektiver Beta2-Agonist, der chemisch als (R*,R*)-(±)-N-[2-Hydroxy-5-[1-Hydroxy-2-[[ 2-(4-Methoxyphenyl)-1-methylethyl]amino]ethyl]phenyl]formamid, (E)-2-Butendioat (2:1), Dihydrat. Die Summenformel von Formoterol ist C42H56N4O14 und sein Molekulargewicht beträgt 840,9. Seine Strukturformel lautet:
Formoterolfumarat-Dihydrat ist ein leicht wasserlösliches Pulver. Sein Octanol-Wasser-Verteilungskoeffizient bei pH 7,4 beträgt 2,6. Der pKa-Wert von Formoterolfumarat-Dihydrat bei 25°C beträgt 7,9 für die Phenolgruppe und 9,2 für die Aminogruppe.
INDIKATIONEN
Behandlung von Asthma
SYMBICORT 4,5 µg ist angezeigt zur Behandlung von Asthma bei Patienten ab 6 Jahren.
SYMBICORT sollte bei Patienten angewendet werden, die mit einem Langzeitmedikament zur Asthmakontrolle, wie z. B. einem inhalativen Kortikosteroid (ICS), nicht ausreichend kontrolliert werden können oder deren Erkrankung den Beginn einer Behandlung mit einem inhalativen Kortikosteroid und einem langwirksamen beta2-adrenergen Agonisten (LABA) rechtfertigt.
Wichtige Nutzungsbeschränkungen
Erhaltungstherapie der chronisch obstruktiven Lungenerkrankung
SYMBICORT 160/4.5 ist indiziert für die Erhaltungstherapie der Atemwegsobstruktion bei Patienten mit chronisch obstruktiver Lungenerkrankung (COPD), einschließlich chronischer Bronchitis und/oder Emphysem. SYMBICORT 160/4.5 ist auch angezeigt, um Exazerbationen von COPD zu reduzieren. SYMBICORT 160/4,5 ist die einzige Stärke, die für die Behandlung von COPD angezeigt ist.
Wichtige Nutzungsbeschränkungen
DOSIERUNG UND ANWENDUNG
Verwaltungsinformationen
SYMBICORT 160 mg sollte als 2 Inhalationen zweimal täglich (morgens und abends im Abstand von etwa 12 Stunden) jeden Tag nur oral inhaliert verabreicht werden. Nach der Inhalation sollte der Patient den Mund mit Wasser ausspülen, ohne zu schlucken.
Bereiten Sie SYMBICORT 4,5 mcg vor der ersten Anwendung vor, indem Sie zwei Testsprühstöße vom Gesicht weg in die Luft geben und vor jedem Sprühstoß 5 Sekunden lang gut schütteln. In Fällen, in denen der Inhalator länger als 7 Tage nicht verwendet wurde oder wenn er fallen gelassen wurde, bereiten Sie den Inhalator erneut vor, indem Sie ihn vor jedem Sprühstoß gut schütteln und zwei Testsprühstöße vom Gesicht weg in die Luft abgeben.
Eine häufigere Verabreichung oder eine größere Anzahl von Inhalationen (mehr als 2 Inhalationen zweimal täglich) der verschriebenen Stärke von SYMBICORT wird nicht empfohlen, da bei manchen Patienten mit höherer Formoterol-Dosierung eher Nebenwirkungen auftreten. Patienten, die SYMBICORT anwenden, sollten aus keinem Grund zusätzliches LABA verwenden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Asthma
Wenn in der Zeit zwischen den Dosen Asthmasymptome auftreten, sollte zur sofortigen Linderung ein inhalativer, kurz wirksamer Beta2-Agonist eingenommen werden.
Erwachsene und jugendliche Patienten ab 12 Jahren
Für Patienten ab 12 Jahren beträgt die Dosierung 2 Inhalationen von SYMBICORT 80/4,5 oder SYMBICORT 160/4,5 zweimal täglich.
Die empfohlenen Anfangsdosierungen für SYMBICORT für Patienten im Alter von 12 Jahren und älter basieren auf dem Schweregrad des Asthmas des Patienten oder dem Grad der Kontrolle der Asthmasymptome und dem Risiko von Exazerbationen unter gegenwärtigen inhalativen Kortikosteroiden.
Die empfohlene Höchstdosis für erwachsene und jugendliche Patienten ab 12 Jahren beträgt SYMBICORT 160/4,5, zwei Inhalationen zweimal täglich.
Eine Verbesserung der Asthmakontrolle nach inhalativer Verabreichung von SYMBICORT kann innerhalb von 15 Minuten nach Beginn der Behandlung eintreten, obwohl der maximale Nutzen möglicherweise erst 2 Wochen oder länger nach Beginn der Behandlung erreicht wird. Einzelne Patienten werden eine unterschiedliche Zeit bis zum Einsetzen und Grad der Symptomlinderung erfahren.
Bei Patienten, die nach 1-2-wöchiger Therapie mit SYMBICORT 80/4,5 nicht ausreichend auf die Anfangsdosis ansprechen, kann die Substitution mit SYMBICORT 160/4,5 eine zusätzliche Asthmakontrolle bewirken.
Wenn ein zuvor wirksames Dosierungsschema von SYMBICORT 4,5 µg keine ausreichende Asthmakontrolle bietet, sollte das therapeutische Schema neu bewertet und zusätzliche therapeutische Optionen (z. B. Ersetzen der niedrigeren Stärke von SYMBICORT 4,5 µg durch die höhere Stärke, Hinzufügen zusätzlicher inhalativer Dosen) empfohlen werden Kortikosteroid oder die Einleitung oraler Kortikosteroide) sollte in Erwägung gezogen werden.
Pädiatrische Patienten im Alter von 6 bis unter 12 Jahren
Für Patienten im Alter von 6 bis unter 12 Jahren beträgt die Dosierung 2 Inhalationen von SYMBICORT 80/4,5 zweimal täglich.
Chronisch obstruktive Lungenerkrankung
Für Patienten mit COPD ist die empfohlene Dosis SYMBICORT 160/4,5, zwei Inhalationen zweimal täglich.
Wenn in der Zeit zwischen den Dosen Kurzatmigkeit auftritt, sollte zur sofortigen Linderung ein inhalativer, kurz wirksamer Beta2-Agonist eingenommen werden.
WIE GELIEFERT
Darreichungsformen und Stärken
SYMBICORT ist als Dosieraerosol mit einer Kombination aus Budesonid (80 oder 160 µg) und Formoterol (4,5 µg) als Inhalationsaerosol in den folgenden zwei Stärken erhältlich: 80/4,5 und 160/4,5. Jede Dosisstärke enthält 60 oder 120 Sprühstöße pro Kanister. Jede Stärke von SYMBICORT 160 mg wird mit einem roten Kunststoffbetätiger mit einer grauen Staubkappe geliefert.
Lagerung und Handhabung
SYMBICORT ist in zwei Stärken erhältlich und wird in folgenden Packungsgrößen geliefert:
Darreichungsformen und Stärken
Jede Stärke wird als druckbeaufschlagter Aluminiumkanister mit aufgesetztem Zählgerät, einem roten Kunststoff-Betätigergehäuse mit weißem Mundstück und aufgesetzter grauer Staubkappe geliefert. Jeder 120er Inhalationsbehälter hat ein Nettofüllgewicht von 10,2 Gramm und jeder 60er Inhalationsbehälter hat ein Nettofüllgewicht von 6,9 Gramm (SYMBICORT 80/4,5) oder 6 Gramm (SYMBICORT 160/4,5). Jeder Kanister ist in einem Folienbeutel mit Trockenmittelbeutel verpackt und in einen Karton gelegt. Jeder Karton enthält einen Kanister und eine Packungsbeilage.
Der SYMBICORT 4,5-mcg-Behälter sollte nur mit dem SYMBICORT 4,5-mcg-Betätiger verwendet werden, und der SYMBICORT-Betätiger sollte nicht mit anderen Arzneimitteln zur Inhalation verwendet werden.
Die richtige Menge des Medikaments bei jeder Inhalation kann nicht gewährleistet werden, nachdem die angegebene Anzahl von Inhalationen aus dem Kanister verwendet wurde, auch wenn sich der Inhalator möglicherweise nicht vollständig leer anfühlt und weiter funktioniert. Der Inhalator sollte entsorgt werden, wenn die angegebene Anzahl von Inhalationen verwendet wurde oder innerhalb von 3 Monaten nach Entnahme aus dem Folienbeutel. Tauchen Sie den Kanister niemals in Wasser, um die verbleibende Menge im Kanister zu bestimmen („Schwimmtest“).
Bei kontrollierter Raumtemperatur von 20 °C bis 25 °C (68 °F bis 77 °F) lagern [siehe USP]. Bewahren Sie den Inhalator mit dem Mundstück nach unten auf.
Für beste Ergebnisse sollte der Kanister vor Gebrauch Raumtemperatur haben. Vor Gebrauch 5 Sekunden gut schütteln.
Von Kindern fern halten. Nicht in die Augen sprühen.
INHALT UNTER DRUCK.
Nicht durchstechen oder verbrennen. Nicht in der Nähe von Hitze oder offenem Feuer lagern. Temperaturen über 120 °F können zum Platzen führen. Behälter niemals ins Feuer oder in die Verbrennungsanlage werfen.
Hergestellt für: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 Von: AstraZeneca Dunkerque Production, Dunkerque, Frankreich. Überarbeitet: Dezember 2017
NEBENWIRKUNGEN
Die Verwendung von LABA kann zu Folgendem führen:
Die systemische und inhalative Anwendung von Kortikosteroiden kann zu Folgendem führen:
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können die in den klinischen Studien zu einem Medikament beobachteten Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien zu einem anderen Medikament verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Erfahrung in klinischen Studien bei Asthma
Erwachsene und jugendliche Patienten ab 12 Jahren
Die Gesamtsicherheitsdaten bei Erwachsenen und Jugendlichen basieren auf 10 aktiv- und placebokontrollierten klinischen Studien, in denen 3393 Patienten ab 12 Jahren (2052 Frauen und 1341 Männer) mit Asthma unterschiedlichen Schweregrades mit SYMBICORT 80/4,5 oder 160 behandelt wurden /4,5 12 bis 52 Wochen lang ein- oder zweimal täglich 2 Inhalationen eingenommen. In diesen Studien waren die Patienten unter SYMBICORT im Mittel 38 Jahre alt und überwiegend Kaukasier (82 %).
Die Häufigkeit häufiger Nebenwirkungen in Tabelle 2 unten basiert auf gepoolten Daten aus drei 12-wöchigen, doppelblinden, placebokontrollierten klinischen Studien, an denen 401 erwachsene und jugendliche Patienten (148 Männer und 253 Frauen) im Alter von 12 Jahren und älter teilnahmen mit 2 Inhalationen von SYMBICORT 80/4,5 oder SYMBICORT 160/4,5 zweimal täglich behandelt. Die SYMBICORT-Gruppe bestand aus überwiegend kaukasischen (84 %) Patienten mit einem Durchschnittsalter von 38 Jahren und einem mittleren Prozentsatz des vorhergesagten FEV1 zu Studienbeginn von 76 bzw. 68 für die Behandlungsgruppen mit 80/4,5 µg bzw. 160/4,5 µg. Kontrollarme zum Vergleich umfassten 2 Inhalationen von Budesonid HFA Dosieraerosol (MDI) 80 oder 160 µg, Formoterol Trockenpulverinhalator (DPI) 4,5 µg oder Placebo (MDI und DPI) zweimal täglich. Tabelle 2 enthält alle unerwünschten Ereignisse, die mit einer Inzidenz von > 3 % in einer beliebigen SYMBICORT-Gruppe und häufiger als in der Placebo-Gruppe bei zweimal täglicher Gabe auftraten. Bei der Berücksichtigung dieser Daten sollte die längere durchschnittliche Dauer der Patientenexposition bei SYMBICORT-Patienten berücksichtigt werden, da die Inzidenzen nicht für ein Ungleichgewicht der Behandlungsdauer angepasst werden.
Langzeitsicherheit – Klinische Studien zu Asthma bei Patienten ab 12 Jahren
Langzeitstudien zur Sicherheit bei jugendlichen und erwachsenen Patienten ab 12 Jahren, die bis zu 1 Jahr lang mit Dosen von bis zu 1280/36 µg/Tag (640/18 µg zweimal täglich) behandelt wurden, zeigten keine klinisch bedeutsamen Änderungen der Inzidenz noch neue Arten von unerwünschten Ereignissen, die nach längerer Behandlungsdauer auftreten. Ebenso wurden bis zu 1 Jahr lang keine signifikanten oder unerwarteten Muster von Anomalien bei Sicherheitsmaßnahmen beobachtet, einschließlich Chemie, Hämatologie, EKG, Holter-Monitor und HPA-Achsen-Bewertungen.
Pädiatrische Patienten im Alter von 6 bis unter 12 Jahren
Die Sicherheitsdaten für pädiatrische Patienten im Alter von 6 bis unter 12 Jahren basieren auf 1 Studie mit einer Behandlungsdauer von 12 Wochen. Patienten (79 Frauen und 105 Männer), die bei Studieneintritt inhalative Kortikosteroide erhielten, wurden randomisiert SYMBICORT 80/4,5 (n = 92) oder Budesonid pMDI 80 mcg (n = 92), 2 Inhalationen zweimal täglich, zugeteilt. Das allgemeine Sicherheitsprofil dieser Patienten war ähnlich dem, das bei Patienten ab 12 Jahren beobachtet wurde, die SYMBICORT 80/4,5 zweimal täglich in Studien mit ähnlichem Design erhielten. Häufige Nebenwirkungen, die bei mit SYMBICORT 80/4,5 behandelten Patienten mit einer Häufigkeit von ≥ 3 % und häufiger auftraten als bei Patienten, die nur mit Budesonid pMDI 80 mcg behandelt wurden, waren Infektionen der oberen Atemwege, Pharyngitis, Kopfschmerzen und Rhinitis.
Erfahrung aus klinischen Studien bei chronisch obstruktiver Lungenerkrankung
Die unten beschriebenen Sicherheitsdaten spiegeln die Exposition gegenüber SYMBICORT 160/4,5 bei 1783 Patienten wider. SYMBICORT 160/4.5 wurde in zwei placebokontrollierten Lungenfunktionsstudien (Dauer 6 und 12 Monate) und zwei aktiv kontrollierten Exazerbationsstudien (Dauer 6 und 12 Monate) bei Patienten mit COPD untersucht.
Die Inzidenz häufiger unerwünschter Ereignisse in Tabelle 3 unten basiert auf gepoolten Daten aus zwei doppelblinden, placebokontrollierten klinischen Studien zur Lungenfunktion (Dauer 6 und 12 Monate), an denen 771 erwachsene COPD-Patienten (496 Männer und 275 Frauen) teilnahmen 40 Jahren und älter wurden mit SYMBICORT 160/4,5 behandelt, zwei Inhalationen zweimal täglich. Von diesen Patienten wurden 651 für 6 Monate und 366 für 12 Monate behandelt. Die SYMBICORT-Gruppe bestand aus überwiegend kaukasischen (93 %) Patienten mit einem mittleren Alter von 63 Jahren und einem mittleren Prozentsatz des vorhergesagten FEV1 zu Studienbeginn von 33 %. Kontrollarme zum Vergleich umfassten 2 Inhalationen von Budesonid HFA (MDI) 160 mcg, Formoterol (DPI) 4,5 mcg oder Placebo (MDI und DPI) zweimal täglich. Tabelle 3 enthält alle unerwünschten Ereignisse, die in der SYMBICORT-Gruppe mit einer Inzidenz von ≥ 3 % und häufiger als in der Placebo-Gruppe auftraten. Bei der Berücksichtigung dieser Daten sollte die längere durchschnittliche Expositionsdauer des Patienten gegenüber SYMBICORT 4,5 µg berücksichtigt werden, da die Inzidenzen nicht für ein Ungleichgewicht der Behandlungsdauer angepasst werden.
Andere Lungeninfektionen als Pneumonie (hauptsächlich Bronchitis) traten bei einem größeren Prozentsatz der mit SYMBICORT 160/4,5 behandelten Patienten im Vergleich zu Placebo auf (7,9 % vs. 5,1 %). Es wurden keine klinisch bedeutsamen oder unerwarteten Muster von Anomalien beobachtet, die bis zu 1 Jahr lang in Chemie, Hämatologie, EKG, EKG (Holter)-Überwachung, HPA-Achse, Knochenmineraldichte und ophthalmologischen Beurteilungen beobachtet wurden.
Die Sicherheitsergebnisse aus den beiden doppelblinden, aktiv kontrollierten Exazerbationsstudien (Dauer 6 und 12 Monate), in denen 1012 erwachsene COPD-Patienten (616 Männer und 396 Frauen) im Alter von 40 Jahren und älter mit SYMBICORT 160/4,5 behandelt wurden, zwei Inhalationen zweimal täglich stimmten mit den Lungenfunktionsstudien überein.
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von SYMBICORT nach der Zulassung festgestellt. Da diese Reaktionen freiwillig aus einer Population unbekannter Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen kausalen Zusammenhang mit der Arzneimittelexposition herzustellen. Einige dieser Nebenwirkungen wurden möglicherweise auch in klinischen Studien mit SYMBICORT beobachtet.
Herzerkrankungen: Angina pectoris, Tachykardie, atriale und ventrikuläre Tachyarrhythmien, Vorhofflimmern, Extrasystolen, Herzklopfen
Endokrine Störungen: Hyperkortizismus, Verringerung der Wachstumsgeschwindigkeit bei pädiatrischen Patienten
Augenerkrankungen: Katarakt, Glaukom, erhöhter Augeninnendruck
Gastrointestinale Störungen: oropharyngeale Candidiasis, Übelkeit
Erkrankungen des Immunsystems: Sofortige und verzögerte Überempfindlichkeitsreaktionen wie anaphylaktische Reaktion, Angioödem, Bronchospasmus, Urtikaria, Exanthem, Dermatitis, Juckreiz
Stoffwechsel- und Ernährungsstörungen: Hyperglykämie, Hypokaliämie
Erkrankungen des Bewegungsapparates, des Bindegewebes und der Knochen: Muskelkrämpfe
Erkrankungen des Nervensystems: Zittern, Schwindel
Psychische Störungen: Verhaltensstörungen, Schlafstörungen, Nervosität, Erregung, Depression, Unruhe
Erkrankungen der Atemwege, des Brustraums und Mediastinums: Dysphonie, Husten, Rachenreizung
Erkrankungen der Haut und des Unterhautzellgewebes: Hautblutungen
Gefäßerkrankungen: Hypotonie, Bluthochdruck
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
In klinischen Studien hat die gleichzeitige Verabreichung von SYMBICORT und anderen Arzneimitteln wie kurzwirksamen Beta2-Agonisten, intranasalen Kortikosteroiden und Antihistaminika/Abschwellmitteln nicht zu einer erhöhten Häufigkeit von Nebenwirkungen geführt. Mit SYMBICORT wurden keine formellen Wechselwirkungsstudien durchgeführt.
Inhibitoren von Cytochrom P4503A4
Der Hauptstoffwechselweg von Kortikosteroiden, einschließlich Budesonid, einem Bestandteil von SYMBICORT, verläuft über das Isoenzym 3A4 (CYP3A4) von Cytochrom P450 (CYP). Nach oraler Verabreichung von Ketoconazol, einem starken Inhibitor von CYP3A4, stieg die mittlere Plasmakonzentration von oral verabreichtem Budesonid an. Die gleichzeitige Verabreichung von CYP3A4 kann den Metabolismus von Budesonid hemmen und die systemische Exposition gegenüber Budesonid erhöhen. Vorsicht ist geboten, wenn die gleichzeitige Anwendung von SYMBICORT 4,5 µg mit Langzeit-Ketoconazol und anderen bekannten starken CYP3A4-Inhibitoren (z. B. Ritonavir, Atazanavir, Clarithromycin, Indinavir, Itraconazol, Nefazodon, Nelfinavir, Saquinavir, Telithromycin) in Betracht gezogen wird [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Monoaminoxidase-Hemmer und trizyklische Antidepressiva
SYMBICORT sollte bei Patienten, die mit Monoaminoxidase-Hemmern oder trizyklischen Antidepressiva behandelt werden, oder innerhalb von 2 Wochen nach Absetzen solcher Arzneimittel mit Vorsicht verabreicht werden, da die Wirkung von Formoterol, einem Bestandteil von SYMBICORT 160 mg, auf das Gefäßsystem durch diese Arzneimittel verstärkt werden kann . In klinischen Studien mit SYMBICORT erhielt eine begrenzte Anzahl von COPD- und Asthmapatienten trizyklische Antidepressiva, weshalb keine klinisch aussagekräftigen Schlussfolgerungen zu unerwünschten Ereignissen gezogen werden können.
Beta-adrenerge Rezeptorblocker
Betablocker (einschließlich Augentropfen) blockieren möglicherweise nicht nur die pulmonale Wirkung von Beta-Agonisten wie Formoterol, einem Bestandteil von SYMBICORT, sondern können bei Patienten mit Asthma schwere Bronchospasmen hervorrufen. Daher sollten Patienten mit Asthma normalerweise nicht mit Betablockern behandelt werden. Unter bestimmten Umständen gibt es jedoch möglicherweise keine akzeptablen Alternativen zur Anwendung von Betarezeptorenblockern bei Patienten mit Asthma. In dieser Situation könnten kardioselektive Betablocker in Betracht gezogen werden, obwohl sie mit Vorsicht verabreicht werden sollten.
Diuretika
Die EKG-Veränderungen und/oder Hypokaliämie, die aus der Verabreichung von nicht kaliumsparenden Diuretika (wie Schleifen- oder Thiaziddiuretika) resultieren können, können durch Beta-Agonisten akut verschlechtert werden, insbesondere wenn die empfohlene Dosis des Beta-Agonisten überschritten wird. Obwohl die klinische Bedeutung dieser Wirkungen nicht bekannt ist, ist bei der gleichzeitigen Verabreichung von SYMBICORT mit nicht kaliumsparenden Diuretika Vorsicht geboten.
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Schwerwiegende Asthma-bezogene Ereignisse – Krankenhausaufenthalte, Intubationen und Tod
Die Anwendung von LABA als Monotherapie (ohne ICS) gegen Asthma ist mit einem erhöhten Asthma-assoziierten Todesrisiko verbunden [siehe Salmeterol Multicenter Asthma Research Trial (SMART)]. Verfügbare Daten aus kontrollierten klinischen Studien deuten auch darauf hin, dass die Anwendung von LABA als Monotherapie das Risiko einer Asthma-bedingten Krankenhauseinweisung bei pädiatrischen und jugendlichen Patienten erhöht. Diese Befunde werden als Klasseneffekt von LABA angesehen. Wenn LABA in Fixdosis-Kombination mit ICS angewendet wird, zeigen Daten aus großen klinischen Studien keinen signifikanten Anstieg des Risikos schwerwiegender Asthma-bedingter Ereignisse (Krankenhauseinweisungen, Intubationen, Tod) im Vergleich zu ICS allein (siehe Schwerwiegende Asthma-bedingte Ereignisse). mit ICS/LABA).
Schwerwiegende Asthma-bezogene Ereignisse mit ICS/LABA
Vier große, 26-wöchige, randomisierte, verblindete, aktiv kontrollierte klinische Sicherheitsstudien wurden durchgeführt, um das Risiko schwerwiegender Asthma-bedingter Ereignisse zu bewerten, wenn LABA in Kombination mit fester Dosis mit ICS im Vergleich zu ICS allein bei Patienten mit Asthma angewendet wurde. Drei Studien schlossen erwachsene und jugendliche Patienten im Alter von ≥ 12 Jahren ein: Eine Studie verglich Budesonid/Formoterol (SYMBICORT) mit Budesonid [siehe Klinische Studien ]; eine Studie verglich Fluticasonpropionat/Salmeterol-Pulver zur Inhalation mit Fluticasonpropionat-Pulver zur Inhalation; und eine Studie verglich Mometasonfuroat/Formoterol mit Mometasonfuroat. Die vierte Studie umfasste pädiatrische Patienten im Alter von 4 bis 11 Jahren und verglich Fluticasonpropionat/Salmeterol-Pulver zur Inhalation mit Fluticasonpropionat-Pulver zur Inhalation. Der primäre Sicherheitsendpunkt für alle vier Studien waren schwerwiegende Asthma-bezogene Ereignisse (Krankenhausaufenthalte, Intubationen und Tod). Ein verblindeter Bewertungsausschuss stellte fest, ob die Ereignisse mit Asthma in Verbindung standen.
Die drei Studien mit Erwachsenen und Jugendlichen wurden so konzipiert, dass eine Risikospanne von 2,0 ausgeschlossen wurde, und die pädiatrische Studie wurde so konzipiert, dass ein Risiko von 2,7 ausgeschlossen wurde. Jede einzelne Studie erreichte ihr vorgegebenes Ziel und zeigte die Nicht-Unterlegenheit von ICS/LABA gegenüber ICS allein. Eine Metaanalyse der drei Studien mit Erwachsenen und Jugendlichen zeigte keinen signifikanten Anstieg des Risikos eines schwerwiegenden Asthma-bedingten Ereignisses mit ICS/LABA-Festdosiskombination im Vergleich zu ICS allein (Tabelle 1). Diese Studien waren nicht darauf ausgelegt, alle Risiken für schwerwiegende Asthma-bedingte Ereignisse mit ICS/LABA im Vergleich zu ICS auszuschließen.
Die pädiatrische Sicherheitsstudie umfasste 6208 pädiatrische Patienten im Alter von 4 bis 11 Jahren, die ICS/LABA (Fluticasonpropionat/Salmeterol-Pulver zur Inhalation) oder ICS (Fluticasonpropionat-Pulver zur Inhalation) erhielten. In dieser Studie erlitten 27/3.107 (0,9 %) Patienten, die zu ICS/LABA randomisiert wurden, und 21/3.101 (0,7 %) Patienten, die zu ICS randomisiert wurden, ein schwerwiegendes Asthma-bedingtes Ereignis. Es gab keine asthmabedingten Todesfälle oder Intubationen. ICS/LABA zeigte kein signifikant erhöhtes Risiko für ein schwerwiegendes Asthma-bezogenes Ereignis im Vergleich zu ICS, basierend auf der vorab festgelegten Risikospanne (2,7), mit einem geschätzten Risikoverhältnis von Zeit bis zum ersten Ereignis von 1,29 (95 % KI: 0,73 , 2.27).
Salmeterol Multizentrische Asthma-Forschungsstudie (SMART)
Eine 28-wöchige, placebokontrollierte US-Studie, in der die Sicherheit von Salmeterol mit Placebo verglichen wurde, jeweils zusätzlich zu einer üblichen Asthmatherapie, zeigte eine Zunahme asthmabedingter Todesfälle bei Patienten, die Salmeterol erhielten (13/13.176 bei mit Salmeterol behandelten Patienten gegenüber 3 /13.179 bei mit Placebo behandelten Patienten; relatives Risiko: 4,37 [95 % KI 1,25; 15,34]). Die Verwendung von Hintergrund-ICS war in SMART nicht erforderlich. Das erhöhte Asthma-bedingte Todesrisiko wird als Klasseneffekt der LABA-Monotherapie angesehen.
Formoterol-Monotherapie-Studien
Klinische Studien mit Formoterol als Monotherapie deuteten auf eine höhere Inzidenz schwerer Asthma-Exazerbationen bei Patienten hin, die Formoterol erhielten, als bei Patienten, die Placebo erhielten. Der Umfang dieser Studien war nicht ausreichend, um den Unterschied bei schwerwiegenden Asthma-Exazerbationen zwischen den Behandlungsgruppen genau zu quantifizieren.
Verschlechterung der Krankheit und akute Episoden
Die Behandlung mit SYMBICORT 4,5 µg sollte bei Patienten während sich schnell verschlechternder oder potenziell lebensbedrohlicher Asthma- oder COPD-Episoden nicht begonnen werden. SYMBICORT wurde bei Patienten mit sich akut verschlechterndem Asthma oder COPD nicht untersucht. Die Einleitung von SYMBICORT 4,5 mcg in dieser Einstellung ist nicht angemessen.
Die zunehmende Anwendung von inhalativen, kurz wirksamen Beta2-Agonisten ist ein Marker für eine Verschlechterung des Asthmas. In dieser Situation benötigt der Patient eine sofortige Neubewertung mit Neubewertung des Behandlungsschemas, wobei besonders berücksichtigt werden muss, ob die derzeitige Stärke von SYMBICORT durch eine höhere Stärke ersetzt, zusätzliches inhalatives Kortikosteroid hinzugefügt oder systemische Kortikosteroide eingeleitet werden müssen. Die Patienten sollten nicht mehr als 2 Inhalationen zweimal täglich (morgens und abends) von SYMBICORT anwenden.
SYMBICORT 160 mg sollte nicht zur Linderung akuter Symptome, dh als Notfalltherapie zur Behandlung akuter Episoden von Bronchospasmen, angewendet werden. Ein inhalativer, kurz wirksamer Beta2-Agonist, nicht SYMBICORT, sollte verwendet werden, um akute Symptome wie Atemnot zu lindern.
Zu Beginn der Behandlung mit SYMBICORT 4,5 µg sollten Patienten, die regelmäßig (z. B. 4-mal täglich) kurz wirkende Beta2-Agonisten oral oder inhalativ eingenommen haben, angewiesen werden, die regelmäßige Einnahme dieser Arzneimittel einzustellen.
Übermäßige Verwendung von SYMBICORT 4,5 mcg und Verwendung mit anderen langwirksamen Beta2-Agonisten
Wie andere inhalative Arzneimittel, die beta2-adrenerge Wirkstoffe enthalten, sollte SYMBICORT 4,5 µg nicht häufiger als empfohlen, in höheren Dosen als empfohlen oder in Verbindung mit anderen Arzneimitteln, die LABA enthalten, angewendet werden, da dies zu einer Überdosierung führen kann. Klinisch signifikante kardiovaskuläre Wirkungen und Todesfälle wurden im Zusammenhang mit der übermäßigen Anwendung von inhalativen Sympathomimetika berichtet. Patienten, die SYMBICORT anwenden, sollten aus keinem Grund ein zusätzliches LABA (z. B. Salmeterol, Formoterolfumarat, Arformoteroltartrat) anwenden, einschließlich der Vorbeugung von belastungsinduziertem Bronchospasmus (EIB) oder der Behandlung von Asthma oder COPD.
Lokale Effekte
In klinischen Studien traten bei Patienten, die mit SYMBICORT behandelt wurden, lokalisierte Infektionen des Mundes und des Rachens mit Candida albicans auf. Wenn sich eine solche Infektion entwickelt, sollte sie mit einer geeigneten lokalen oder systemischen (dh oralen Antimykotika) Therapie behandelt werden, während die Behandlung mit SYMBICORT 160 mg fortgesetzt wird, aber manchmal muss die Therapie mit SYMBICORT 4,5 µg unterbrochen werden. Weisen Sie den Patienten an, seinen Mund nach der Inhalation mit Wasser auszuspülen, ohne es herunterzuschlucken, um das Risiko einer oropharyngealen Candidiasis zu verringern.
Lungenentzündung und andere Infektionen der unteren Atemwege
Ärzte sollten bei Patienten mit COPD auf die mögliche Entwicklung einer Lungenentzündung achten, da sich die klinischen Merkmale von Lungenentzündung und Exazerbationen häufig überschneiden. Nach der inhalativen Verabreichung von Kortikosteroiden wurde über Infektionen der unteren Atemwege, einschließlich Lungenentzündung, berichtet.
In einer 6-monatigen Lungenfunktionsstudie mit 1704 COPD-Patienten gab es eine höhere Inzidenz von anderen Lungeninfektionen als Pneumonie (z. B. Bronchitis, virale Infektionen der unteren Atemwege usw.) bei Patienten, die SYMBICORT 160/4,5 erhielten (7,6 %). als bei denen, die SYMBICORT 80/4,5 (3,2 %), Formoterol 4,5 µg (4,6 %) oder Placebo (3,3 %) erhielten. Pneumonien traten in der SYMBICORT 160/4,5-Gruppe (1,1 %) im Vergleich zu Placebo (1,3 %) nicht häufiger auf. In einer 12-monatigen Lungenfunktionsstudie mit 1964 COPD-Patienten traten bei Patienten, die SYMBICORT 160/4,5 erhielten (8,1 %), auch andere Lungeninfektionen als Pneumonie häufiger auf als bei Patienten, die SYMBICORT 80/4,5 (6,9 %) erhielten. Formoterol 4,5 µg (7,1 %) oder Placebo (6,2 %). Ähnlich wie in der 6-monatigen Studie trat eine Pneumonie nicht häufiger in der SYMBICORT 160/4,5-Gruppe (4,0 %) im Vergleich zu Placebo (5,0 %) auf.
Immunsuppression
Patienten, die Medikamente einnehmen, die das Immunsystem unterdrücken, sind anfälliger für Infektionen als gesunde Personen. Windpocken und Masern beispielsweise können bei empfindlichen Kindern oder Erwachsenen, die Kortikosteroide einnehmen, einen schwereren oder sogar tödlichen Verlauf nehmen. Bei solchen Kindern oder Erwachsenen, die diese Krankheiten nicht hatten oder ordnungsgemäß geimpft wurden, sollte besonders darauf geachtet werden, eine Exposition zu vermeiden. Wie sich Dosis, Art und Dauer der Kortikosteroidverabreichung auf das Risiko der Entwicklung einer disseminierten Infektion auswirken, ist nicht bekannt. Der Beitrag der zugrunde liegenden Erkrankung und/oder einer vorherigen Behandlung mit Kortikosteroiden zum Risiko ist ebenfalls nicht bekannt. Bei Exposition kann gegebenenfalls eine Therapie mit Varizella-Zoster-Immunglobulin (VZIG) oder gepooltem intravenösen Immunglobulin (IVIG) angezeigt sein. Bei Exposition gegenüber Masern kann eine Prophylaxe mit gepooltem intramuskulärem Immunglobulin (IG) angezeigt sein (siehe die jeweiligen Packungsbeilagen für vollständige VZIG- und IG-Verschreibungsinformationen). Wenn sich Windpocken entwickeln, kann eine Behandlung mit antiviralen Mitteln in Betracht gezogen werden. Die Immunantwort auf Varizellen-Impfstoff wurde bei pädiatrischen Patienten mit Asthma im Alter von 12 Monaten bis 8 Jahren mit Budesonid-Inhalationssuspension untersucht.
Eine offene, nicht randomisierte klinische Studie untersuchte die Immunantwort auf Varizellen-Impfstoff bei 243 Asthmapatienten im Alter von 12 Monaten bis 8 Jahren, die mit Budesonid-Inhalationssuspension 0,25 mg bis 1 mg täglich (n = 151) oder einer nicht-kortikosteroidischen Asthmatherapie (n =92) (dh Beta2-Agonisten, Leukotrienrezeptorantagonisten, Cromone). Der Prozentsatz der Patienten, die als Reaktion auf die Impfung einen seroprotektiven Antikörpertiter von > 5,0 (gpELISA-Wert) entwickelten, war ähnlich bei Patienten, die mit Budesonid-Inhalationssuspension behandelt wurden (85 %), im Vergleich zu Patienten, die mit einer Asthmatherapie ohne Kortikosteroide behandelt wurden (90 %). Kein Patient, der mit Budesonid-Inhalationssuspension behandelt wurde, entwickelte infolge der Impfung Windpocken.
Inhalative Kortikosteroide sollten bei Patienten mit aktiven oder ruhenden Tuberkuloseinfektionen der Atemwege, wenn überhaupt, mit Vorsicht angewendet werden; unbehandelte systemische Pilz-, Bakterien-, Virus- oder Parasiteninfektionen; oder okulärer Herpes simplex.
Überführung von Patienten aus der systemischen Kortikosteroidtherapie
Besondere Vorsicht ist bei Patienten geboten, die von systemisch wirksamen Kortikosteroiden auf inhalative Kortikosteroide umgestellt wurden, da bei Patienten mit Asthma während und nach der Umstellung von systemischen Kortikosteroiden auf weniger systemisch verfügbare inhalative Kortikosteroide Todesfälle aufgrund von Nebenniereninsuffizienz aufgetreten sind. Nach dem Absetzen von systemischen Kortikosteroiden dauert es einige Monate, bis sich die Hypothalamus-Hypophysen-Nebennierenfunktion (HPA) erholt hat.
Patienten, die zuvor mit 20 mg oder mehr Prednison (oder einem Äquivalent) pro Tag behandelt wurden, sind möglicherweise am anfälligsten, insbesondere wenn ihre systemischen Kortikosteroide fast vollständig abgesetzt wurden. Während dieser Phase der HPA-Unterdrückung können Patienten Anzeichen und Symptome einer Nebenniereninsuffizienz zeigen, wenn sie einem Trauma, einer Operation oder einer Infektion (insbesondere Gastroenteritis) oder anderen Erkrankungen ausgesetzt sind, die mit einem schweren Elektrolytverlust einhergehen. Obwohl SYMBICORT während dieser Episoden zur Kontrolle der Asthmasymptome führen kann, liefert es in den empfohlenen Dosen systemisch weniger als die normalen physiologischen Mengen an Glucocorticoid und liefert NICHT die Mineralocorticoid-Aktivität, die zur Bewältigung dieser Notfälle erforderlich ist.
In Zeiten von Stress, einem schweren Asthmaanfall oder einer schweren COPD-Exazerbation sollten Patienten, die von systemischen Kortikosteroiden abgesetzt wurden, angewiesen werden, die oralen Kortikosteroide (in großen Dosen) sofort wieder aufzunehmen und sich für weitere Anweisungen an ihren Arzt zu wenden. Diese Patienten sollten auch angewiesen werden, einen Warnhinweis mit sich zu führen, der darauf hinweist, dass sie in Stressphasen, bei einem schweren Asthmaanfall oder bei einer schweren COPD-Exazerbation möglicherweise zusätzliche systemische Kortikosteroide benötigen.
Patienten, die orale Kortikosteroide benötigen, sollten nach der Umstellung auf SYMBICORT langsam von der Anwendung systemischer Kortikosteroide entwöhnt werden. Eine Prednison-Reduktion kann erreicht werden, indem die tägliche Prednison-Dosis während der Therapie mit SYMBICORT wöchentlich um 2,5 mg reduziert wird. Während des Absetzens von oralen Kortikosteroiden sollten die Lungenfunktion (mittleres forciertes exspiratorisches Volumen in 1 Sekunde [FEV1] oder morgendlicher maximaler expiratorischer Fluss [PEF]), die Anwendung von Beta-Agonisten und Asthma- oder COPD-Symptome sorgfältig überwacht werden. Darüber hinaus sollten Patienten auf Anzeichen und Symptome einer Nebenniereninsuffizienz wie Müdigkeit, Abgeschlagenheit, Schwäche, Übelkeit und Erbrechen sowie Hypotonie beobachtet werden.
Die Umstellung von Patienten von einer systemischen Kortikosteroidtherapie auf inhalative Kortikosteroide oder SYMBICORT kann Zustände demaskieren, die zuvor durch die systemische Kortikosteroidtherapie unterdrückt wurden (z. B. Rhinitis, Konjunktivitis, Ekzem, Arthritis, eosinophile Erkrankungen). Bei manchen Patienten können trotz Aufrechterhaltung oder sogar Verbesserung der Atemfunktion Symptome eines Entzugs von systemisch wirksamen Kortikosteroiden auftreten (z. B. Gelenk- und/oder Muskelschmerzen, Abgeschlagenheit, Depression).
Hyperkortizismus und Nebennierensuppression
Budesonid, ein Bestandteil von SYMBICORT, hilft häufig bei der Kontrolle von Asthma- und COPD-Symptomen mit einer geringeren Unterdrückung der HPA-Funktion als therapeutisch äquivalente orale Dosen von Prednison. Da Budesonid in den Kreislauf aufgenommen wird und in höheren Dosen systemisch aktiv sein kann, sind die vorteilhaften Wirkungen von SYMBICORT zur Minimierung der HPA-Dysfunktion nur dann zu erwarten, wenn die empfohlenen Dosierungen nicht überschritten werden und einzelne Patienten auf die niedrigste wirksame Dosis titriert werden.
Wegen der Möglichkeit einer systemischen Resorption von inhalativen Kortikosteroiden sollten Patienten, die mit SYMBICORT 160 mg behandelt werden, sorgfältig auf Hinweise auf systemische Kortikosteroidwirkungen beobachtet werden. Besondere Sorgfalt sollte bei der Beobachtung von Patienten postoperativ oder während Stressphasen auf Anzeichen einer unzureichenden Nebennierenreaktion angewendet werden.
Es ist möglich, dass bei einer kleinen Anzahl von Patienten systemische Kortikosteroidwirkungen wie Hyperkortizismus und Nebennierensuppression (einschließlich Nebennierenkrise) auftreten, insbesondere wenn Budesonid über längere Zeiträume in höheren als den empfohlenen Dosen verabreicht wird. Wenn solche Wirkungen auftreten, sollte die Dosierung von SYMBICORT langsam reduziert werden, im Einklang mit anerkannten Verfahren zur Reduzierung systemischer Kortikosteroide und zur Behandlung von Asthmasymptomen.
Arzneimittelwechselwirkungen mit starken Cytochrom P450 3A4-Inhibitoren
Vorsicht ist geboten, wenn die gleichzeitige Anwendung von SYMBICORT 4,5 µg mit Ketoconazol und anderen bekannten starken CYP3A4-Hemmern (z. B. Ritonavir, Atazanavir, Clarithromycin, Indinavir, Itraconazol, Nefazodon, Nelfinavir, Saquinavir, Telithromycin) in Betracht gezogen wird, da Nebenwirkungen mit einer erhöhten systemischen Exposition zusammenhängen zu Budesonid kann auftreten [vgl WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ].
Paradoxer Bronchospasmus und Symptome der oberen Atemwege
Wie andere inhalative Medikamente kann SYMBICORT 160 mg paradoxe Bronchospasmen hervorrufen, die lebensbedrohlich sein können. Wenn nach der Gabe von SYMBICORT 160 mg ein paradoxer Bronchospasmus auftritt, sollte dieser sofort mit einem inhalativen, kurzwirksamen Bronchodilatator behandelt werden, SYMBICORT sollte sofort abgesetzt und eine alternative Therapie eingeleitet werden.
Sofortige Überempfindlichkeitsreaktionen
Unmittelbare Überempfindlichkeitsreaktionen können nach Verabreichung von SYMBICORT 160 mg auftreten, wie Fälle von Urtikaria, Angioödem, Hautausschlag und Bronchospasmus zeigen.
Auswirkungen auf das Herz-Kreislauf-System und das zentrale Nervensystem
Übermäßige beta-adrenerge Stimulation wurde mit Krampfanfällen, Angina pectoris, Bluthochdruck oder Hypotonie, Tachykardie mit Frequenzen von bis zu 200 Schlägen/min, Arrhythmien, Nervosität, Kopfschmerzen, Zittern, Herzklopfen, Übelkeit, Schwindel, Müdigkeit, Unwohlsein und Schlaflosigkeit in Verbindung gebracht [siehe ÜBERDOSIERUNG ]. Daher sollte SYMBICORT 160 mg, wie alle Produkte, die sympathomimetische Amine enthalten, bei Patienten mit Herz-Kreislauf-Erkrankungen, insbesondere Koronarinsuffizienz, Herzrhythmusstörungen und Bluthochdruck, mit Vorsicht angewendet werden.
Formoterol, ein Bestandteil von SYMBICORT, kann bei manchen Patienten einen klinisch signifikanten kardiovaskulären Effekt hervorrufen, gemessen anhand der Pulsfrequenz, des Blutdrucks und/oder der Symptome. Obwohl solche Wirkungen nach Verabreichung von Formoterol in den empfohlenen Dosen gelegentlich auftreten, muss das Arzneimittel möglicherweise abgesetzt werden, wenn sie auftreten. Darüber hinaus wurde von Beta-Agonisten berichtet, dass sie EKG-Veränderungen wie eine Abflachung der T-Welle, eine Verlängerung des QTc-Intervalls und eine ST-Streckensenkung hervorrufen. Die klinische Bedeutung dieser Befunde ist nicht bekannt. Todesfälle wurden im Zusammenhang mit dem übermäßigen Gebrauch von inhalativen Sympathomimetika berichtet.
Verringerung der Knochenmineraldichte
Bei Langzeitanwendung von Produkten, die inhalative Kortikosteroide enthalten, wurde eine Abnahme der Knochenmineraldichte (BMD) beobachtet. Die klinische Bedeutung kleiner BMD-Veränderungen im Hinblick auf Langzeitfolgen wie Fraktur ist unbekannt. Patienten mit wichtigen Risikofaktoren für einen verringerten Knochenmineralgehalt, wie z. B. längere Immobilisierung, Osteoporose in der Familienanamnese, postmenopausaler Status, Tabakkonsum, fortgeschrittenes Alter, schlechte Ernährung oder chronische Einnahme von Arzneimitteln, die die Knochenmasse reduzieren können (z. B. Antikonvulsiva, orale Kortikosteroide). ) sollten überwacht und mit etablierten Behandlungsstandards behandelt werden. Da Patienten mit COPD häufig mehrere Risikofaktoren für eine reduzierte BMD haben, wird eine Beurteilung der BMD vor Beginn der Behandlung mit SYMBICORT 160 mg und danach in regelmäßigen Abständen empfohlen. Wenn eine signifikante Reduzierung der BMD beobachtet wird und SYMBICORT immer noch als medizinisch wichtig für die COPD-Therapie dieses Patienten angesehen wird, sollte die Verwendung von Medikamenten zur Behandlung oder Vorbeugung von Osteoporose dringend in Betracht gezogen werden.
Die Auswirkungen der Behandlung mit SYMBICORT 160/4,5, SYMBICORT 80/4,5, Formoterol 4,5 µg oder Placebo auf die BMD wurden bei einer Untergruppe von 326 Patienten (Frauen und Männer im Alter von 41 bis 88 Jahren) mit COPD in der 12-Monats-Lungenfunktion untersucht lernen. BMD-Bewertungen der Hüft- und Lendenwirbelsäulenregionen wurden zu Studienbeginn und nach 52 Wochen unter Verwendung von Dual-Energy-Röntgen-Absorptiometrie (DEXA)-Scans durchgeführt. Die mittleren Veränderungen der BMD vom Ausgangswert bis zum Ende der Behandlung waren gering (mittlere Veränderungen reichten von -0,01 bis 0,01 g/cm²). Die ANCOVA-Ergebnisse für die BMD der gesamten Wirbelsäule und der gesamten Hüfte, basierend auf dem Zeitpunkt am Ende der Behandlung, zeigten, dass alle geometrischen LS-Mittelwerte für die paarweisen Vergleiche der Behandlungsgruppen nahe bei 1 lagen, was darauf hindeutet, dass die Gesamt-BMD für die gesamten Hüft- und gesamten Wirbelsäulenregionen für die 12 -Monats-Zeitpunkt waren über den gesamten Behandlungszeitraum stabil.
Auswirkung auf das Wachstum
Oral inhalierte Kortikosteroide können bei der Verabreichung an pädiatrische Patienten zu einer Verringerung der Wachstumsgeschwindigkeit führen. Überwachen Sie das Wachstum von pädiatrischen Patienten, die SYMBICORT routinemäßig erhalten (z. B. mittels Stadiometrie). Um die systemischen Wirkungen von oral inhalativen Kortikosteroiden, einschließlich SYMBICORT, zu minimieren, titrieren Sie die Dosis jedes Patienten auf die niedrigste Dosis, die seine/ihre Symptome wirksam kontrolliert [siehe DOSIERUNG UND ANWENDUNG und Verwendung in bestimmten Bevölkerungsgruppen ].
Glaukom und Katarakte
Glaukom, erhöhter Augeninnendruck und Katarakte wurden bei Patienten mit Asthma und COPD nach Langzeitanwendung von inhalativen Kortikosteroiden, einschließlich Budesonid, einem Bestandteil von SYMBICORT, berichtet. Daher ist eine engmaschige Überwachung bei Patienten mit Sehstörungen oder erhöhtem Augeninnendruck in der Anamnese, Glaukom und/oder Katarakt angezeigt.
Die Auswirkungen der Behandlung mit SYMBICORT 160/4,5, SYMBICORT 80/4,5, Formoterol 4,5 µg oder Placebo auf die Entwicklung von Katarakt oder Glaukom wurden in einer Untergruppe von 461 Patienten mit COPD in der 12-monatigen Lungenfunktionsstudie untersucht. Augenärztliche Untersuchungen wurden zu Studienbeginn, nach 24 Wochen und nach 52 Wochen durchgeführt. Es gab 26 Probanden (6 %) mit einem Anstieg des posterioren subkapsulären Scores vom Ausgangswert bis zum Maximalwert (> 0,7) während des randomisierten Behandlungszeitraums. Veränderungen der posterioren subkapsulären Scores von > 0,7 vom Ausgangswert bis zum Behandlungsmaximum traten bei 11 Patienten (9,0 %) in der SYMBICORT 160/4,5-Gruppe, 4 Patienten (3,8 %) in der SYMBICORT 80/4,5-Gruppe und 5 Patienten (4,2 %) auf der Formoterol-Gruppe und 6 Patienten (5,2 %) in der Placebo-Gruppe.
Eosinophile Zustände und Churg-Strauss-Syndrom
In seltenen Fällen können sich Patienten unter inhalativen Kortikosteroiden mit systemischen eosinophilen Erkrankungen vorstellen. Einige dieser Patienten weisen klinische Merkmale einer Vaskulitis auf, die mit dem Churg-Strauss-Syndrom übereinstimmen, einer Erkrankung, die häufig mit einer systemischen Kortikosteroidtherapie behandelt wird. Diese Ereignisse wurden in der Regel, aber nicht immer, mit der Reduzierung und/oder dem Absetzen der oralen Kortikosteroidtherapie nach der Einführung inhalativer Kortikosteroide in Verbindung gebracht. Ärzte sollten bei ihren Patienten auf Eosinophilie, vaskulitischen Hautausschlag, sich verschlechternde Lungensymptome, kardiale Komplikationen und/oder Neuropathie achten. Ein kausaler Zusammenhang zwischen Budesonid und diesen Grunderkrankungen wurde nicht hergestellt.
Koexistenzbedingungen
SYMBICORT 4,5 mcg sollte wie alle Medikamente, die sympathomimetische Amine enthalten, bei Patienten mit Krampfanfällen oder Thyreotoxikose und bei Patienten, die ungewöhnlich auf sympathomimetische Amine ansprechen, mit Vorsicht angewendet werden. Es wurde berichtet, dass Dosen des verwandten Beta2-Adrenozeptor-Agonisten Albuterol bei intravenöser Verabreichung einen vorbestehenden Diabetes mellitus und eine Ketoazidose verschlimmern.
Hypokaliämie und Hyperglykämie
Beta-adrenerge Agonisten-Medikamente können bei manchen Patienten eine signifikante Hypokaliämie hervorrufen, möglicherweise durch intrazellulären Shunt, der potenziell nachteilige kardiovaskuläre Wirkungen hervorrufen kann [siehe KLINISCHE PHARMAKOLOGIE ]. Die Abnahme des Serumkaliums ist normalerweise vorübergehend und erfordert keine Supplementierung. Klinisch signifikante Veränderungen des Blutzuckers und/oder des Serumkaliums wurden in klinischen Studien mit SYMBICORT in den empfohlenen Dosierungen selten beobachtet.
Informationen zur Patientenberatung
Weisen Sie den Patienten an, die von der FDA zugelassene Patientenkennzeichnung ( PATIENTENINFORMATION und Gebrauchsanweisung ).
Schwerwiegende Ereignisse im Zusammenhang mit Asthma
Informieren Sie Patienten mit Asthma darüber, dass LABA bei alleiniger Anwendung das Risiko einer asthmabedingten Krankenhauseinweisung oder eines asthmabedingten Todes erhöht. Verfügbare Daten zeigen, dass bei gemeinsamer Anwendung von ICS und LABA, wie z. B. mit SYMBICORT, das Risiko für diese Ereignisse nicht signifikant erhöht wird.
Nicht für akute Symptome
Informieren Sie die Patienten darüber, dass SYMBICORT 160 mg nicht dazu bestimmt ist, akute Symptome von Asthma oder COPD zu lindern, und dass zu diesem Zweck keine zusätzlichen Dosen verwendet werden sollten. Raten Sie den Patienten, akute Symptome mit einem inhalativen, kurz wirksamen Beta2-Agonisten wie Albuterol zu behandeln. Versorgen Sie Patienten mit solchen Medikamenten und weisen Sie den Patienten in die Anwendung ein.
Weisen Sie die Patienten an, sofort einen Arzt aufzusuchen, wenn bei ihnen eines der folgenden Symptome auftritt:
Sagen Sie den Patienten, dass sie die Therapie mit SYMBICORT nicht ohne ärztliche/ärztliche Anleitung abbrechen sollten, da die Symptome nach dem Absetzen wieder auftreten können.
Verwenden Sie keine zusätzlichen langwirksamen Beta2-Agonisten
Weisen Sie die Patienten an, keine anderen LABA für Asthma und COPD zu verwenden.
Lokale Effekte
Informieren Sie die Patienten darüber, dass bei einigen Patienten lokalisierte Infektionen mit Candida albicans im Mund und Rachen aufgetreten sind. Wenn sich eine oropharyngeale Candidiasis entwickelt, sollte sie mit einer geeigneten lokalen oder systemischen (dh oralen) Antimykotikatherapie behandelt werden, während die Therapie mit SYMBICORT 4,5 µg fortgesetzt wird, aber manchmal muss die Therapie mit SYMBICORT unter engmaschiger ärztlicher Überwachung vorübergehend unterbrochen werden. Es wird empfohlen, den Mund nach der Inhalation mit Wasser zu spülen, ohne ihn herunterzuschlucken, um das Risiko einer Soorinfektion zu verringern.
Lungenentzündung
Patienten mit COPD haben ein höheres Lungenentzündungsrisiko; Weisen Sie sie an, sich an ihren Arzt zu wenden, wenn sie Symptome einer Lungenentzündung entwickeln.
Immunsuppression
Warnen Sie Patienten, die immunsuppressive Dosen von Kortikosteroiden erhalten, davor, sich Windpocken oder Masern auszusetzen und im Falle einer Exposition unverzüglich ihren Arzt aufzusuchen. Informieren Sie die Patienten über eine mögliche Verschlechterung bestehender Tuberkulose, Pilz-, Bakterien-, Virus- oder Parasiteninfektionen oder Herpes simplex am Auge.
Hyperkortizismus und Nebennierensuppression
Weisen Sie die Patienten darauf hin, dass SYMBICORT systemische kortikosteroide Wirkungen von Hyperkortizismus und Nebennierensuppression verursachen kann. Informieren Sie die Patienten außerdem darüber, dass Todesfälle aufgrund von Nebenniereninsuffizienz während und nach der Umstellung von systemischen Kortikosteroiden aufgetreten sind. Patienten sollten systemische Kortikosteroide langsam ausschleichen, wenn sie auf SYMBICORT umgestellt werden.
Verringerung der Knochenmineraldichte
Weisen Sie Patienten mit einem erhöhten Risiko für eine verringerte BMD darauf hin, dass die Anwendung von Kortikosteroiden ein zusätzliches Risiko darstellen kann.
Reduzierte Wachstumsgeschwindigkeit
Informieren Sie die Patienten darüber, dass oral inhalierte Kortikosteroide, ein Bestandteil von SYMBICORT, bei pädiatrischen Patienten eine Verringerung der Wachstumsgeschwindigkeit verursachen können. Ärzte sollten das Wachstum von Kindern und Jugendlichen, die Kortikosteroide auf beliebige Weise einnehmen, genau beobachten.
Okulare Wirkungen
Die Langzeitanwendung von inhalativen Kortikosteroiden kann das Risiko einiger Augenprobleme (Katarakt oder Glaukom) erhöhen; erwägen Sie regelmäßige Augenuntersuchungen.
Risiken im Zusammenhang mit der Beta-Agonisten-Therapie
Informieren Sie die Patienten über Nebenwirkungen im Zusammenhang mit Beta2-Agonisten, wie z. B. Herzklopfen, Brustschmerzen, schneller Herzschlag, Zittern oder Nervosität.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Budesonid
Zur Bewertung des kanzerogenen Potenzials von Budesonid wurden Langzeitstudien an Ratten und Mäusen mit oraler Verabreichung durchgeführt.
In einer 2-jährigen Studie an Sprague-Dawley-Ratten verursachte Budesonid bei männlichen Ratten bei einer oralen Dosis von 50 µg/kg (entspricht ungefähr der MRHDID bei Erwachsenen und Kindern unter einer µg/m²-Dosis) einen statistisch signifikanten Anstieg der Inzidenz von Gliomen Basis). Bei männlichen und weiblichen Ratten wurde bei oralen Dosen von bis zu 25 bzw. 50 µg/kg (ungefähr äquivalent zu der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) keine Tumorigenität beobachtet. In zwei weiteren 2-Jahres-Studien an männlichen Fischer- und Sprague-Dawley-Ratten verursachte Budesonid bei einer oralen Dosis von 50 µg/kg (ungefähr äquivalent zu der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) keine Gliome. Bei männlichen Sprague-Dawley-Ratten verursachte Budesonid jedoch bei einer oralen Dosis von 50 µg/kg (entspricht etwa der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) einen statistisch signifikanten Anstieg der Inzidenz hepatozellulärer Tumore. Die gleichzeitigen Referenzkortikosteroide (Prednisolon und Triamcinolonacetonid) in diesen beiden Studien zeigten ähnliche Ergebnisse.
In einer 91-wöchigen Studie an Mäusen verursachte Budesonid bei oralen Dosen von bis zu 200 µg/kg (etwa das 2-fache der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) keine behandlungsbedingte Karzinogenität.
Budesonid war in sechs verschiedenen Testsystemen nicht mutagen oder klastogen: Ames-Salmonella/Mikrosomenplattentest, Maus-Mikrokerntest, Maus-Lymphomtest, Chromosomenaberrationstest in menschlichen Lymphozyten, geschlechtsgebundener rezessiver Letaltest in Drosophila melanogaster und DNA-Reparaturanalyse in Ratten Hepatozytenkultur.
Fertilität und Reproduktionsleistung wurden bei Ratten bei subkutanen Dosen von bis zu 80 µg/kg (ungefähr gleich der MRHDID auf µg/m²-Basis) nicht beeinträchtigt. Bei subkutanen Dosen von 20 µg/kg und mehr (weniger als die MRHDID auf µg/m²-Basis) führte es jedoch zu einer Abnahme der pränatalen Lebensfähigkeit und der Lebensfähigkeit der Jungtiere bei der Geburt und während der Laktation, zusammen mit einer Abnahme der Körpergewichtszunahme des Muttertiers ). Bei 5 µg/kg (weniger als der MRHDID auf µg/m²-Basis) wurden keine derartigen Wirkungen festgestellt.
Formoterol
Zur Bewertung des kanzerogenen Potenzials von Formoterolfumarat wurden Langzeitstudien an Mäusen mit oraler Verabreichung und Ratten mit Inhalationsverabreichung durchgeführt.
In einer 24-monatigen Kanzerogenitätsstudie an CD-1-Mäusen verursachte Formoterol in oralen Dosen von 100 µg/kg und mehr (ungefähr das 30- bzw. 15-fache der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) eine Dosis- damit verbundene Zunahme der Inzidenz von Uterus-Leiomyomen.
In einer 24-monatigen Karzinogenitätsstudie an Sprague-Dawley-Ratten wurde eine erhöhte Inzidenz von mesovarischen Leiomyomen und uterinen Leiomyosarkomen bei einer inhalierten Dosis von 130 µg/kg (ungefähr das 70- bzw mcg/m²-Basis). Bei 22 µg/kg (etwa das 12- bzw. 6-fache der MRHDID bei Erwachsenen und Kindern auf µg/m²-Basis) wurden keine Tumore beobachtet.
Andere Beta-Agonisten haben in ähnlicher Weise eine Zunahme von Leiomyomen des Genitaltrakts bei weiblichen Nagetieren gezeigt. Die Relevanz dieser Ergebnisse für die Anwendung beim Menschen ist nicht bekannt.
Formoterol war im Ames-Salmonella/Mikrosomen-Plattentest, im Maus-Lymphom-Test, im Chromosomenaberrationstest in menschlichen Lymphozyten und im Ratten-Mikrokerntest nicht mutagen oder klastogen.
Bei männlichen Ratten, die mit Formoterol in einer oralen Dosis von 15.000 µg/kg (etwa das 2200-fache der MRHDID auf AUC-Basis) behandelt wurden, wurde eine Verringerung der Fertilität und/oder Fortpflanzungsfähigkeit festgestellt. Bei 3000 mcg/kg (etwa das 1600-fache der MRHDID auf mcg/m²-Basis) wurde keine solche Wirkung beobachtet. In einer separaten Studie mit männlichen Ratten, die mit einer oralen Dosis von 15.000 mcg/kg (ungefähr das 8000-fache der MRHDID auf mcg/m²-Basis) behandelt wurden, gab es Befunde von testikulärer tubulärer Atrophie und Spermatrümmern in den Hoden und Oligospermie in den Nebenhoden. Bei weiblichen Ratten wurde bei Dosen bis zu 15.000 mcg/kg (etwa das 1100-fache der MRHDID auf AUC-Basis) keine Wirkung auf die Fertilität festgestellt.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Zusammenfassung der Risiken
Es liegen keine angemessenen und gut kontrollierten Studien zu SYMBICORT 160 mg oder einem seiner Einzelbestandteile, Formoterolfumarat, bei Schwangeren vor; für den anderen Bestandteil Budesonid liegen jedoch Studien vor. In Reproduktionsstudien an Tieren wirkte SYMBICORT 4,5 µg, verabreicht durch Inhalation, teratogen, embryozid und reduzierte das fötale Gewicht bei Ratten bei weniger als der maximal empfohlenen täglichen Inhalationsdosis beim Menschen (MRHDID) auf µg/m²-Basis. Budesonid allein, subkutan verabreicht, war teratogen, embryozid und reduzierte das fötale Gewicht bei Ratten und Kaninchen bei weniger als der MRHDID, aber diese Wirkungen wurden nicht bei Ratten beobachtet, die inhalierte Dosen bis zum 4-fachen der MRHDID erhielten. Studien an schwangeren Frauen haben nicht gezeigt, dass inhaliertes Budesonid allein das Risiko von Anomalien erhöht, wenn es während der Schwangerschaft verabreicht wird. Erfahrungen mit oralen Kortikosteroiden legen nahe, dass Nagetiere anfälliger für teratogene Wirkungen einer Kortikosteroid-Exposition sind als Menschen. Formoterolfumarat allein, oral verabreicht, war bei Ratten und Kaninchen beim 1600- bzw. 65.000-fachen der MRHDID teratogen. Formoterolfumarat war auch embryozid, erhöhte den Verlust der Jungtiere bei der Geburt und während der Laktation und verringerte das Gewicht der Jungtiere bei Ratten beim 110-fachen der MRHDID. Diese Nebenwirkungen traten im Allgemeinen bei großen Vielfachen der MRHDID auf, wenn Formoterolfumarat oral verabreicht wurde, um hohe systemische Expositionen zu erreichen. Bei Ratten, die Inhalationsdosen bis zum 375-fachen der MRHDID erhielten, wurden keine teratogenen, embryoziden oder entwicklungsschädigenden Wirkungen beobachtet.
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten der angegebenen Populationen ist unbekannt. In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwere Geburtsfehler und Fehlgeburten bei klinisch erkannten Schwangerschaften bei 2 % bis 4 % bzw. 15 % bis 20 %.
Klinische Überlegungen
Krankheitsbedingtes mütterliches und/oder embryonale/fetales Risiko
Bei Frauen mit schlecht oder mäßig kontrolliertem Asthma besteht ein erhöhtes Risiko für verschiedene perinatale Nebenwirkungen wie Präeklampsie bei der Mutter und Frühgeburtlichkeit, niedriges Geburtsgewicht und geringes Gestationsalter beim Neugeborenen. Schwangere Frauen mit Asthma sollten engmaschig überwacht und die Medikation nach Bedarf angepasst werden, um eine optimale Asthmakontrolle aufrechtzuerhalten.
Arbeit oder Lieferung
Es gibt keine gut kontrollierten Studien am Menschen, die die Auswirkungen von SYMBICORT 4,5 mcg während der Wehen und der Geburt untersucht haben. Wegen der möglichen Beeinträchtigung der Uteruskontraktilität durch Beta-Agonisten sollte die Anwendung von SYMBICORT 4,5 µg während der Wehen auf Patientinnen beschränkt werden, bei denen der Nutzen das Risiko eindeutig überwiegt.
Daten
Menschliche Daten
Studien an schwangeren Frauen haben nicht gezeigt, dass inhaliertes Budesonid das Risiko von Anomalien erhöht, wenn es während der Schwangerschaft verabreicht wird. Die Ergebnisse einer großen bevölkerungsbasierten prospektiven epidemiologischen Kohortenstudie, in der Daten aus drei schwedischen Registern überprüft wurden, die etwa 99 % der Schwangerschaften von 1995-1997 abdecken (dh schwedisches medizinisches Geburtsregister; Register für angeborene Fehlbildungen; Register für Kinderkardiologie), weisen auf kein erhöhtes Risiko hin bei angeborenen Fehlbildungen durch die Anwendung von inhalativem Budesonid während der Frühschwangerschaft. Angeborene Fehlbildungen wurden 2014 bei Säuglingen untersucht, die von Müttern geboren wurden, die über die Anwendung von inhalativem Budesonid gegen Asthma in der Frühschwangerschaft (normalerweise 10–12 Wochen nach der letzten Monatsblutung) berichteten, dem Zeitraum, in dem die meisten größeren Organfehlbildungen auftreten. Die Rate der registrierten angeborenen Fehlbildungen war im Vergleich zur allgemeinen Bevölkerungsrate ähnlich (3,8 % vs. 3,5 %). Darüber hinaus war die Zahl der Säuglinge, die mit orofazialen Spalten geboren wurden, nach Exposition gegenüber inhaliertem Budesonid ähnlich der erwarteten Zahl in der Normalbevölkerung (4 Kinder vs. 3,3).
Dieselben Daten wurden in einer zweiten Studie verwendet, die die Gesamtzahl auf 2534 Säuglinge brachte, deren Mütter inhaliertem Budesonid ausgesetzt waren. In dieser Studie unterschied sich die Rate angeborener Missbildungen bei Säuglingen, deren Mütter während der Frühschwangerschaft inhalativem Budesonid ausgesetzt waren, nicht von der Rate aller Neugeborenen im gleichen Zeitraum (3,6 %).
Tierdaten
SYMBIKORT
In einer Studie zur embryofetalen Entwicklung bei trächtigen Ratten, denen während der Organogenese vom 6. bis 16. Trächtigkeitstag verabreicht wurde, verursachte SYMBICORT 160 mg Nabelbrüche bei Föten bei Dosen unterhalb der MRHDID (auf mcg/m²-Basis bei maternal inhalierten Dosen von 12/ 0,66 mcg/kg/Tag und mehr). Das fötale Gewicht war um etwa das 5- bzw. 3-fache der MRHDID reduziert (auf AUC-Basis bei einer inhalierten Dosis der Mutter von 80/4,4 µg/kg (Budesonid/Formoterol)). Bei Dosen unterhalb der MRHDID (auf mcg/m²-Basis bei einer von der Mutter inhalierten Dosis von 2,5/0,14 mcg/kg/Tag) wurden keine teratogenen oder embryoziden Wirkungen festgestellt.
Budesonid
In einer Fertilitäts- und Reproduktionsstudie wurde männlichen Ratten 9 Wochen lang und weiblichen 2 Wochen lang vor der Paarung und während der Paarungszeit eine subkutane Dosis verabreicht. Weibchen wurden bis zum Absetzen ihrer Nachkommen dosiert. Budesonid verursachte bei Dosen unterhalb der MRHDID (auf mcg/m²-Basis bei subkutanen Dosen von 20 mcg/ kg/Tag und mehr). Bei einer Dosis unterhalb der MRHDID (auf Mikrogramm/m²-Basis bei einer mütterlichen subkutanen Dosis von 5 Mikrogramm/kg/Tag) wurden keine derartigen Wirkungen festgestellt.
In einer Studie zur embryofetalen Entwicklung bei trächtigen Kaninchen, denen während der Organogenese vom 6. bis 18. Trächtigkeitstag verabreicht wurde, führte Budesonid bei Dosen unterhalb der MRHDID (auf mcg/m²-Basis bei a mütterliche subkutane Dosis von 25 mcg/kg/Tag). In einer Studie zur embryofetalen Entwicklung an trächtigen Ratten, denen während der Organogenese vom 6. bis 15. Trächtigkeitstag eine Dosis verabreicht wurde, verursachte Budesonid ähnliche nachteilige fetale Wirkungen bei Dosen, die etwa dem 8-fachen der MRHDID (auf mcg/m²-Basis bei einer maternalen subkutanen Dosis von 500 µg/kg/Tag). In einer anderen Studie zur embryofetalen Entwicklung bei trächtigen Ratten wurden keine teratogenen oder embryoziden Wirkungen bei Dosen bis zum 4-Fachen der MRHDID (auf einer mcg/m²-Basis bei maternalen Inhalationsdosen von bis zu 250 mcg/kg/Tag) beobachtet.
In einer peri- und postnatalen Entwicklungsstudie mit Ratten, denen vom 15. Tag der Trächtigkeit bis zum 21. Tag nach der Geburt eine Dosis verabreicht wurde, hatte Budesonid keine Auswirkungen auf die Geburt, hatte jedoch eine Auswirkung auf das Wachstum und die Entwicklung der Nachkommen. Bei Dosen unterhalb der MRHDID und höher (auf mcg/m²-Basis bei mütterlichen subkutanen Dosen von 20 mcg/kg/Tag und höher) war die Überlebensrate der Nachkommen verringert und das mittlere Körpergewicht der überlebenden Nachkommen war bei der Geburt und während der Laktation verringert. Diese Befunde traten in Gegenwart maternaler Toxizität auf.
Formoterol
In einer Fertilitäts- und Reproduktionsstudie erhielten männliche Ratten 9 Wochen lang und weibliche 2 Wochen lang vor der Paarung und während der Paarungszeit eine orale Dosis. Weibchen wurden entweder bis zum 19. Trächtigkeitstag oder bis zum Absetzen ihrer Nachkommen dosiert. Männchen wurden bis zu 25 Wochen dosiert. Nabelbrüche wurden bei Rattenföten bei oralen Dosen beobachtet, die das 1600-fache und mehr der MRHDID (auf mcg/m²-Basis bei maternalen oralen Dosen von 3000 mcg/kg/Tag und höher) betrugen. Brachygnathie wurde bei Rattenföten bei einer Dosis beobachtet, die das 8000-fache der MRHDID betrug (auf mcg/m²-Basis bei einer maternalen oralen Dosis von 15.000 mcg/kg/Tag). Die Schwangerschaft wurde bei einer Dosis verlängert, die das 8000-fache der MRHDID betrug (auf Mikrogramm/m²-Basis bei einer mütterlichen oralen Dosis von 15.000 Mikrogramm/kg/Tag). Todesfälle bei Föten und Jungtieren traten während der Trächtigkeit bei Dosen auf, die etwa dem 1600-Fachen der MRHDID und höher (auf einer mcg/m²-Basis bei oralen Dosen von 3000 mcg/kg/Tag und höher) entsprachen.
In einer Studie zur embryofetalen Entwicklung bei trächtigen Ratten, denen während der Organogenese vom 6. bis 15. Trächtigkeitstag eine Dosis verabreicht wurde, wurden bei Dosen bis zum 375-Fachen der MRHDID (auf mcg/m²-Basis bei Inhalation durch die Mutter) keine teratogenen, embryoziden oder entwicklungsschädigenden Wirkungen beobachtet Dosen bis zu 690 mcg/kg/Tag).
In einer Studie zur embryofetalen Entwicklung bei trächtigen Kaninchen, denen während der Organogenese vom 6. bis 18. Trächtigkeitstag eine Dosis verabreicht wurde, wurden subkapsuläre Zysten auf der Leber bei den Föten bei einer Dosis beobachtet, die dem 65.000-fachen der MRHDID (auf mcg/m²-Basis bei einer mütterlichen Dosis) entsprach orale Dosis von 60.000 mcg/kg/Tag). Bei Dosen bis zum 3800-fachen der MRHDID (auf mcg/m²-Basis bei maternalen oralen Dosen von bis zu 3500 mcg/kg/Tag) wurden keine teratogenen Wirkungen beobachtet.
In einer prä- und postnatalen Entwicklungsstudie erhielten trächtige weibliche Ratten Formoterol in oralen Dosen von 0, 210, 840 und 3400 mcg/kg/Tag vom 6. Trächtigkeitstag bis zur Laktation. Die Überlebensrate der Jungtiere war von der Geburt bis zum 26. Tag nach der Geburt bei Dosen, die das 110-fache der MRHDID und höher betrugen (auf mcg/m²-Basis bei maternalen oralen Dosen von 210 mcg/kg/Tag und höher), obwohl es keine Hinweise auf eine Dosis-Wirkungs-Beziehung gab Beziehung. Es gab keine behandlungsbedingten Auswirkungen auf die körperliche, funktionelle und Verhaltensentwicklung von Rattenwelpen.
Stillzeit
Zusammenfassung der Risiken
Es liegen keine Daten zu den Wirkungen von SYMBICORT 4,5 µg, Budesonid oder Formoterolfumarat auf das gestillte Kind oder auf die Milchproduktion vor. Budesonid ist wie andere inhalative Kortikosteroide in der Muttermilch vorhanden [siehe Daten ]. Es liegen keine Daten zum Vorhandensein von Formoterolfumarat in der Muttermilch vor. Formoterolfumarat ist in Rattenmilch vorhanden [vgl Daten ]. Die Entwicklungs- und Gesundheitsvorteile des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an SYMBICORT 4,5 µg und möglichen Nebenwirkungen von SYMBICORT 160 mg auf den gestillten Säugling oder aufgrund des zugrunde liegenden Zustands der Mutter berücksichtigt werden.
Daten
Humandaten mit Budesonid, das über einen Trockenpulverinhalator verabreicht wird, weisen darauf hin, dass die gesamte tägliche orale Dosis von Budesonid, die dem Säugling in der Muttermilch zur Verfügung steht, etwa 0,3 % bis 1 % der von der Mutter inhalierten Dosis beträgt [siehe KLINISCHE PHARMAKOLOGIE ]. Für SYMBICORT 4,5 µg ist die Budesonid-Dosis, die dem Säugling in der Muttermilch als Prozentsatz der mütterlichen Dosis zur Verfügung steht, erwartungsgemäß ähnlich.
In der Fertilitäts- und Reproduktionsstudie an Ratten wurden die Plasmaspiegel von Formoterol bei Jungtieren am 15. Tag nach der Geburt gemessen [vgl Verwendung in bestimmten Bevölkerungsgruppen ]. Es wurde geschätzt, dass die maximale Plasmakonzentration, die die Jungtiere vom mütterlichen Tier bei der höchsten Dosis von 15 mg/kg nach dem Stillen erhielten, 4,4 % betrug (0,24 nmol/l für einen Wurf gegenüber 5,5 nmol/l für die Mutter). .
Pädiatrische Verwendung
Die Sicherheit und Wirksamkeit von SYMBICORT bei Asthmapatienten ab 12 Jahren wurde in Studien mit einer Dauer von bis zu 12 Monaten nachgewiesen. In den beiden 12-wöchigen, doppelblinden, placebokontrollierten Zulassungsstudien in den USA wurden 25 Patienten im Alter von 12 bis 17 Jahren zweimal täglich mit SYMBICORT behandelt [siehe Klinische Studien ]. Die Wirksamkeitsergebnisse in dieser Altersgruppe waren ähnlich denen, die bei Patienten ab 18 Jahren beobachtet wurden. Es gab keine offensichtlichen Unterschiede in der Art oder Häufigkeit der berichteten Nebenwirkungen in dieser Altersgruppe im Vergleich zu Patienten ab 18 Jahren.
Die Sicherheit und Wirksamkeit von SYMBICORT 80/4,5 bei Asthmapatienten im Alter von 6 bis unter 12 Jahren wurde in Studien mit einer Dauer von bis zu 12 Wochen nachgewiesen [siehe Klinische Studien ]. Das Sicherheitsprofil bei diesen Patienten entsprach dem bei Patienten ab 12 Jahren, die ebenfalls SYMBICORT erhielten [siehe NEBENWIRKUNGEN ].
Die Sicherheit und Wirksamkeit von SYMBICORT 160 mg bei Asthmapatienten unter 6 Jahren ist nicht erwiesen.
Kontrollierte klinische Studien haben gezeigt, dass oral inhalative Kortikosteroide, einschließlich Budesonid, ein Bestandteil von SYMBICORT, eine Verringerung der Wachstumsgeschwindigkeit bei pädiatrischen Patienten verursachen können. Dieser Effekt wurde in Abwesenheit von Labornachweisen für eine Unterdrückung der HPA-Achse beobachtet, was darauf hindeutet, dass die Wachstumsgeschwindigkeit ein empfindlicherer Indikator für die systemische Kortikosteroid-Exposition bei pädiatrischen Patienten ist als einige häufig verwendete Tests der HPA-Achsenfunktion. Die Langzeitwirkung dieser Verringerung der Wachstumsgeschwindigkeit im Zusammenhang mit oral inhalierten Kortikosteroiden, einschließlich der Auswirkungen auf die endgültige Körpergröße, ist nicht bekannt. Das Potenzial für ein „aufholendes“ Wachstum nach Absetzen der Behandlung mit oral inhalativen Kortikosteroiden wurde nicht ausreichend untersucht.
In einer Studie mit asthmatischen Kindern im Alter von 5 bis 12 Jahren hatten diejenigen, die zweimal täglich 200 mcg Budesonid DPI (n = 311) erhielten, am Ende eines Jahres eine 1,1-cm-Wachstumsminderung im Vergleich zu denen, die Placebo erhielten (n = 418). ; der Unterschied zwischen diesen beiden Behandlungsgruppen nahm über drei Jahre zusätzlicher Behandlung nicht weiter zu. Am Ende von 4 Jahren hatten Kinder, die mit Budesonid DPI behandelt wurden, und Kinder, die mit Placebo behandelt wurden, ähnliche Wachstumsgeschwindigkeiten. Die aus dieser Studie gezogenen Schlussfolgerungen können durch die ungleiche Verwendung von Kortikosteroiden in den Behandlungsgruppen und die Einbeziehung von Daten von Patienten, die im Verlauf der Studie die Pubertät erreichten, verfälscht werden.
Das Wachstum von pädiatrischen Patienten, die oral inhalative Kortikosteroide, einschließlich SYMBICORT, erhalten, sollte überwacht werden. Wenn bei einem Kind oder Jugendlichen, das Kortikosteroide einnimmt, eine Wachstumshemmung auftritt, sollte die Möglichkeit in Betracht gezogen werden, dass er/sie besonders empfindlich auf diese Wirkung reagiert. Die potenziellen Wachstumseffekte einer längeren Behandlung sollten gegen den erzielten klinischen Nutzen abgewogen werden. Um die systemischen Wirkungen von oral inhalativen Kortikosteroiden, einschließlich SYMBICORT, zu minimieren, sollte jeder Patient auf die niedrigste Stärke titriert werden, die sein Asthma wirksam kontrolliert [siehe DOSIERUNG UND ANWENDUNG ].
Geriatrische Verwendung
Von der Gesamtzahl der Asthmapatienten, die in zwei 12-wöchigen Studien und einer 26-wöchigen Postmarketing-Studie zweimal täglich mit SYMBICORT 160 mg behandelt wurden, waren 791 65 Jahre oder älter, davon 141 75 Jahre oder älter.
In den COPD-Studien mit einer Dauer von 6 bis 12 Monaten waren 810 Patienten, die mit SYMBICORT 160/4,5 behandelt wurden, zwei Inhalationen zweimal täglich, 65 Jahre und älter, und davon waren 177 Patienten 75 Jahre und älter. Insgesamt wurden keine Unterschiede in der Sicherheit oder Wirksamkeit zwischen diesen Patienten und jüngeren Patienten beobachtet, und andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt.
Wie bei anderen Produkten, die Beta2-Agonisten enthalten, ist bei der Anwendung von SYMBICORT bei geriatrischen Patienten mit gleichzeitiger kardiovaskulärer Erkrankung, die durch Beta2-Agonisten beeinträchtigt werden könnte, besondere Vorsicht geboten.
Basierend auf den verfügbaren Daten für SYMBICORT oder seine Wirkstoffe ist keine Anpassung der Dosierung von SYMBICORT 4,5 µg bei geriatrischen Patienten gerechtfertigt.
Leberfunktionsstörung
Formale pharmakokinetische Studien mit SYMBICORT wurden bei Patienten mit eingeschränkter Leberfunktion nicht durchgeführt. Da jedoch sowohl Budesonid als auch Formoterolfumarat überwiegend durch Leberstoffwechsel ausgeschieden werden, kann eine Beeinträchtigung der Leberfunktion zu einer Akkumulation von Budesonid und Formoterolfumarat im Plasma führen. Daher sollten Patienten mit Lebererkrankungen engmaschig überwacht werden.
Nierenfunktionsstörung
Formale pharmakokinetische Studien mit SYMBICORT wurden bei Patienten mit eingeschränkter Nierenfunktion nicht durchgeführt.
ÜBERDOSIS
SYMBIKORT
SYMBICORT enthält sowohl Budesonid als auch Formoterol; daher gelten für SYMBICORT die nachfolgend beschriebenen Risiken einer Überdosierung der einzelnen Bestandteile. In pharmakokinetischen Studien wurden Patienten mit COPD Einzeldosen von 960/54 Mikrogramm (12 Sprühstöße SYMBICORT 80/4,5) und 1280/36 Mikrogramm (8 Sprühstöße 160/4,5) verabreicht. Insgesamt 1920/54 Mikrogramm (12 Sprühstöße von SYMBICORT 160/4,5) wurden als Einzeldosis sowohl gesunden Probanden als auch Patienten mit Asthma verabreicht. In einer aktiv kontrollierten Langzeitstudie zur Sicherheit bei jugendlichen und erwachsenen Asthmapatienten ab 12 Jahren wurde SYMBICORT 160/4.5 bis zu 12 Monate lang in Dosen bis zum Doppelten der höchsten empfohlenen Tagesdosis verabreicht. In keiner dieser Studien wurden klinisch signifikante Nebenwirkungen beobachtet.
Budesonid
Das Potenzial für akute toxische Wirkungen nach einer Überdosierung von Budesonid ist gering. Bei Anwendung überhöhter Dosen über einen längeren Zeitraum können systemische Kortikosteroidwirkungen wie Hyperkortizismus auftreten [vgl WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Budesonid, das fünfmal so hoch wie die empfohlene Höchstdosis (3200 mcg täglich) über 6 Wochen an Menschen verabreicht wurde, verursachte eine signifikante Verringerung (27 %) der Cortisolreaktion im Plasma auf eine 6-stündige ACTH-Infusion im Vergleich zu Placebo (+1 %). Die entsprechende Wirkung von 10 mg Prednison täglich war eine 35 %ige Reduktion der Plasma-Cortisol-Reaktion auf ACTH.
Formoterol
Eine Überdosierung von Formoterol würde wahrscheinlich zu einer Verstärkung der für Beta2-Agonisten typischen Wirkungen führen: Krampfanfälle, Angina pectoris, Bluthochdruck, Hypotonie, Tachykardie, atriale und ventrikuläre Tachyarrhythmien, Nervosität, Kopfschmerzen, Zittern, Herzklopfen, Muskelkrämpfe, Übelkeit, Schwindel, Schlafstörungen , metabolische Azidose, Hyperglykämie, Hypokaliämie. Wie bei allen sympathomimetischen Medikamenten kann der Missbrauch von Formoterol zu Herzstillstand und sogar zum Tod führen. Es wurden keine klinisch signifikanten Nebenwirkungen beobachtet, wenn Formoterol erwachsenen Patienten mit akuter Bronchokonstriktion in einer Dosis von 90 Mikrogramm/Tag über 3 Stunden oder stabilen Asthmatikern dreimal täglich in einer Gesamtdosis von 54 Mikrogramm/Tag über 3 Tage verabreicht wurde.
Die Behandlung einer Formoterol-Überdosierung besteht aus dem Absetzen des Medikaments zusammen mit der Einleitung einer geeigneten symptomatischen und/oder unterstützenden Therapie. Die vernünftige Anwendung eines kardioselektiven Betarezeptorenblockers kann in Betracht gezogen werden, wobei zu berücksichtigen ist, dass solche Medikamente Bronchospasmen hervorrufen können. Es gibt keine ausreichenden Beweise, um festzustellen, ob eine Dialyse bei einer Überdosierung von Formoterol vorteilhaft ist. Im Falle einer Überdosierung wird eine Herzüberwachung empfohlen.
KONTRAINDIKATIONEN
Die Anwendung von SYMBICORT ist unter folgenden Bedingungen kontraindiziert:
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
SYMBIKORT
SYMBICORT enthält sowohl Budesonid als auch Formoterol; daher gelten für SYMBICORT die nachfolgend für die einzelnen Komponenten beschriebenen Wirkmechanismen. Diese Medikamente stellen zwei Klassen von Medikamenten dar (ein synthetisches Kortikosteroid und ein langwirksamer selektiver Beta2-Adrenozeptor-Agonist), die unterschiedliche Wirkungen auf die klinischen, physiologischen und entzündlichen Indizes von COPD und Asthma haben.
Budesonid
Budesonid ist ein entzündungshemmendes Corticosteroid, das eine starke Glucocorticoid-Aktivität und eine schwache Mineralocorticoid-Aktivität aufweist. In standardmäßigen In-vitro- und Tiermodellen hat Budesonid eine etwa 200-fach höhere Affinität zum Glukokortikoidrezeptor und eine 1000-fach höhere topische entzündungshemmende Wirkung als Cortisol (Ratten-Crotonöl-Ohrödem-Assay). Als Maß für die systemische Aktivität ist Budesonid 40-mal wirksamer als Cortisol, wenn es subkutan verabreicht wird, und 25-mal wirksamer, wenn es oral verabreicht wird, im Ratten-Thymus-Involutions-Assay.
In Glucocorticoid-Rezeptor-Affinitätsstudien war die 22R-Form von Budesonid zweimal so aktiv wie das 22S-Epimer. In-vitro-Studien zeigten, dass sich die beiden Formen von Budesonid nicht ineinander umwandeln.
Entzündung ist eine wichtige Komponente in der Pathogenese von COPD und Asthma. Kortikosteroide haben ein breites Spektrum an inhibitorischen Aktivitäten gegen mehrere Zelltypen (z. B. Mastzellen, Eosinophile, Neutrophile, Makrophagen und Lymphozyten) und Mediatoren (z. B. Histamin, Eicosanoide, Leukotriene und Zytokine), die an allergischen und nicht-allergischen Reaktionen beteiligt sind Entzündung. Diese entzündungshemmenden Wirkungen von Kortikosteroiden können zu ihrer Wirksamkeit bei COPD und Asthma beitragen.
Studien an Asthmapatienten haben ein günstiges Verhältnis zwischen topischer entzündungshemmender Aktivität und systemischer Kortikosteroidwirkung über einen weiten Dosisbereich von Budesonid gezeigt. Dies wird durch eine Kombination aus einer relativ hohen lokalen entzündungshemmenden Wirkung, einem ausgedehnten hepatischen First-Pass-Abbau des oral absorbierten Arzneimittels (85 %–95 %) und der geringen Potenz der gebildeten Metaboliten erklärt.
Formoterol
Formoterolfumarat ist ein langwirksamer selektiver beta2-adrenerger Agonist (beta2-Agonist) mit schnellem Wirkungseintritt. Inhaliertes Formoterolfumarat wirkt lokal in der Lunge als Bronchodilatator. In-vitro-Studien haben gezeigt, dass Formoterol eine mehr als 200-mal stärkere agonistische Aktivität an Beta2-Rezeptoren als an Beta1-Rezeptoren hat. Die In-vitro-Bindungsselektivität für Beta2- gegenüber Beta1-Adrenozeptoren ist für Formoterol höher als für Albuterol (5-mal), während Salmeterol ein höheres (3-mal) Beta2-Selektivitätsverhältnis als Formoterol aufweist.
Obwohl Beta2-Rezeptoren die vorherrschenden adrenergen Rezeptoren in der glatten Muskulatur der Bronchien und Beta1-Rezeptoren die vorherrschenden Rezeptoren im Herzen sind, gibt es auch Beta2-Rezeptoren im menschlichen Herzen, die 10 % bis 50 % der gesamten Beta-adrenergen Rezeptoren ausmachen. Die genaue Funktion dieser Rezeptoren ist nicht bekannt, aber sie lassen die Möglichkeit vermuten, dass sogar hochselektive Beta2-Agonisten kardiale Wirkungen haben können.
Die pharmakologischen Wirkungen von Beta2-Adrenozeptor-Agonisten, einschließlich Formoterol, sind zumindest teilweise auf die Stimulierung der intrazellulären Adenylcyclase zurückzuführen, dem Enzym, das die Umwandlung von Adenosintriphosphat (ATP) in zyklisches 3',5'-Adenosinmonophosphat katalysiert ( zyklisches AMP). Erhöhte zyklische AMP-Spiegel bewirken eine Entspannung der glatten Bronchialmuskulatur und eine Hemmung der Freisetzung von Mediatoren der unmittelbaren Überempfindlichkeit aus Zellen, insbesondere aus Mastzellen.
In-vitro-Tests zeigen, dass Formoterol die Freisetzung von Mastzellmediatoren wie Histamin und Leukotrienen aus der menschlichen Lunge hemmt. Formoterol hemmt auch die Histamin-induzierte Plasmaalbumin-Extravasation bei anästhesierten Meerschweinchen und hemmt den Allergen-induzierten Einstrom von Eosinophilen bei Hunden mit Hyperreaktivität der Atemwege. Die Relevanz dieser In-vitro- und Tierbefunde für den Menschen ist nicht bekannt.
Pharmakodynamik
Asthma
Kardiovaskuläre Wirkungen
In einer Einzeldosis-Crossover-Studie mit 201 Patienten mit persistierendem Asthma wurden Einzeldosisbehandlungen mit 4,5, 9 und 18 Mikrogramm Formoterol in Kombination mit 320 Mikrogramm Budesonid, verabreicht über SYMBICORT, mit Budesonid 320 Mikrogramm allein verglichen. Im Vergleich zu Budesonid wurden dosisabhängige Verbesserungen des FEV1 nachgewiesen. EKGs und Blutproben für Glukose und Kalium wurden nach der Dosis erhalten. Bei SYMBICORT 4,5 µg wurden bei steigenden Formoterol-Dosen im Vergleich zu Budesonid geringfügige mittlere Anstiege der Serumglukose und Abnahmen des Serumkaliums (+0,44 mmol/l bzw. -0,18 mmol/l bei der höchsten Dosis) beobachtet. In EKGs führte SYMBICORT im Vergleich zu Budesonid allein zu einer geringen dosisabhängigen mittleren Erhöhung der Herzfrequenz (ungefähr 3 bpm bei der höchsten Dosis) und der QTc-Intervalle (3-6 ms). Kein Proband hatte einen QT- oder QTc-Wert ≥500 ms.
In den Vereinigten Staaten wurden in fünf 12-wöchigen aktiv- und placebokontrollierten Studien und einer 6-monatigen aktiv-kontrollierten Studie 2976 Patienten mit Asthma im Alter von 6 Jahren und älter untersucht. Die systemischen pharmakodynamischen Wirkungen von Formoterol (Herz-/Pulsfrequenz, Blutdruck, QTc-Intervall, Kalium und Glukose) waren bei mit SYMBICORT behandelten Patienten ähnlich, verglichen mit Patienten, die mit Formoterol-Trockeninhalationspulver 4,5 µg, 2 Inhalationen zweimal täglich, behandelt wurden. Kein Patient hatte während der Behandlung einen QT- oder QTc-Wert von ≥ 500 ms.
In drei placebokontrollierten Studien mit Jugendlichen und Erwachsenen mit Asthma ab 12 Jahren wurde bei insgesamt 1232 Patienten (553 Patienten in der SYMBICORT 160 mg-Gruppe) eine auswertbare kontinuierliche 24-Stunden-EKG-Überwachung durchgeführt. Insgesamt gab es in der Gruppe mit SYMBICORT 160 mg im Vergleich zu Placebo keine wesentlichen Unterschiede beim Auftreten von ventrikulärer oder supraventrikulärer Ektopie und keinen Hinweis auf ein erhöhtes Risiko für klinisch signifikante Rhythmusstörungen.
HPA-Achsen-Effekte
Insgesamt wurden bei mit SYMBICORT behandelten erwachsenen oder jugendlichen Patienten in Dosen von bis zu 640/18 µg/Tag im Vergleich zu Budesonid keine klinisch bedeutsamen Wirkungen auf die HPA-Achse, gemessen anhand von 24-Stunden-Cortisol im Urin, beobachtet.
Chronisch obstruktive Lungenerkrankung
Kardiovaskuläre Wirkungen
In zwei COPD-Lungenfunktionsstudien mit einer Dauer von 6 Monaten und 12 Monaten mit 3668 COPD-Patienten wurden keine klinisch bedeutsamen Unterschiede bei Pulsfrequenz, Blutdruck, Kalium und Glukose zwischen SYMBICORT, den einzelnen Bestandteilen von SYMBICORT 160 mg und Placebo festgestellt [siehe Klinische Studien ].
EKGs, die bei mehreren Klinikbesuchen während der Behandlung in beiden Studien aufgezeichnet wurden, zeigten keine klinisch bedeutsamen Unterschiede hinsichtlich Herzfrequenz, PR-Intervall, QRS-Dauer, Herzfrequenz, Anzeichen einer kardialen Ischämie oder Arrhythmien zwischen SYMBICORT 160/4,5 als Monopräparat und Placebo, die alle als 2 Inhalationen verabreicht wurden zweimal täglich. Basierend auf den EKGs trat bei 6 Patienten, die mit SYMBICORT 160/4,5 behandelt wurden, 6 Patienten, die mit Formoterol 4,5 µg behandelt wurden, und 6 Patienten in der Placebogruppe ein Vorhofflimmern oder -flattern auf, das zu Studienbeginn nicht vorhanden war. Es gab keine Fälle von nicht anhaltender ventrikulärer Tachykardie in den Gruppen mit SYMBICORT 160/4,5, Formoterol 4,5 µg oder Placebo.
In der 12-monatigen Studie wurde bei 520 Patienten eine auswertbare kontinuierliche 24-Stunden-EKG-Überwachung (Holter) vor der ersten Dosis und nach ungefähr 1 und 4 Behandlungsmonaten durchgeführt. Es wurden keine klinisch bedeutsamen Unterschiede bei ventrikulären oder supraventrikulären Arrhythmien, ventrikulären oder supraventrikulären ektopischen Schlägen oder der Herzfrequenz zwischen den Gruppen beobachtet, die mit SYMBICORT 160/4,5, Formoterol oder Placebo, eingenommen als 2 Inhalationen zweimal täglich, behandelt wurden. Basierend auf der EKG (Holter)-Überwachung kam es bei einem Patienten unter SYMBICORT 160/4,5, bei keinem Patienten unter Formoterol 4,5 mcg und bei drei Patienten in der Placebogruppe zu Vorhofflimmern oder -flattern, das zu Studienbeginn nicht vorhanden war.
HPA-Achsen-Effekte
Bei einer gepoolten Untergruppe (n = 616) von Patienten aus zwei COPD-Lungenfunktionsstudien wurden 24-Stunden-Urinkortisolmessungen durchgeführt. Die Daten zeigten ungefähr 30 % niedrigere mittlere 24-Stunden-Werte von freiem Cortisol im Urin nach chronischer Verabreichung (> 6 Monate) von SYMBICORT im Vergleich zu Placebo. SYMBICORT 4,5 µg zeigte eine vergleichbare Cortisolsuppression wie Budesonid 160 µg allein oder die gleichzeitige Gabe von Budesonid 160 µg und Formoterol 4,5 µg. Bei Patienten, die bis zu 12 Monate lang mit SYMBICORT 160 mg oder Placebo behandelt wurden, war der Prozentsatz der Patienten, die bei dieser Messung von normal auf niedrig wechselten, im Allgemeinen vergleichbar.
Andere Budesonid-Produkte
Um zu bestätigen, dass die systemische Absorption kein signifikanter Faktor für die klinische Wirksamkeit von inhaliertem Budesonid ist, wurde eine klinische Studie an Patienten mit Asthma durchgeführt, in der 400 µg Budesonid, das über einen unter Druck stehenden Dosieraerosol mit einem Abstandshalter verabreicht wurde, mit 1400 µg oralem Budesonid und Placebo verglichen wurden . Die Studie zeigte trotz vergleichbarer systemischer Spiegel die Wirksamkeit von inhaliertem Budesonid, nicht aber von oral eingenommenem Budesonid. Somit wird die therapeutische Wirkung herkömmlicher Dosen von oral inhaliertem Budesonid weitgehend durch seine direkte Wirkung auf die Atemwege erklärt.
Es wurde gezeigt, dass inhaliertes Budesonid die Reaktivität der Atemwege gegenüber verschiedenen Provokationsmodellen, einschließlich Histamin, Methacholin, Natriummetabisulfit und Adenosinmonophosphat, bei Patienten mit hyperreaktiven Atemwegen verringert. Die klinische Relevanz dieser Modelle ist nicht sicher.
Die Vorbehandlung mit inhaliertem Budesonid, 1600 µg täglich (800 µg zweimal täglich) über 2 Wochen reduzierte die akute (Frühphasenreaktion) und verzögerte (Spätphasenreaktion) Abnahme des FEV1 nach inhalativer Allergenprovokation.
Die systemischen Wirkungen von inhalativen Kortikosteroiden hängen mit der systemischen Exposition gegenüber solchen Arzneimitteln zusammen. Pharmakokinetische Studien haben gezeigt, dass sowohl bei Erwachsenen als auch bei Kindern mit Asthma die systemische Exposition gegenüber Budesonid bei SYMBICORT 160 mg niedriger ist als bei inhaliertem Budesonid, das in der gleichen verabreichten Dosis über einen Trockenpulverinhalator verabreicht wird [siehe KLINISCHE PHARMAKOLOGIE ]. Daher ist zu erwarten, dass die systemischen Wirkungen (HPA-Achse und Wachstum) von Budesonid, das von SYMBICORT abgegeben wird, nicht größer sind als die, die für inhalatives Budesonid berichtet werden, wenn es in vergleichbaren Dosen über den Trockenpulverinhalator verabreicht wird [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
HPA-Achsen-Effekte
Die Wirkung von inhaliertem Budesonid, das über einen Trockenpulverinhalator verabreicht wurde, auf die HPA-Achse wurde bei 905 Erwachsenen und 404 pädiatrischen Patienten mit Asthma untersucht. Bei den meisten Patienten blieb die Fähigkeit, die Cortisolproduktion als Reaktion auf Stress zu erhöhen, wie durch den Cosyntropin (ACTH)-Stimulationstest festgestellt, bei der Behandlung mit Budesonid in den empfohlenen Dosen erhalten. Bei erwachsenen Patienten, die mit 100, 200, 400 oder 800 µg zweimal täglich über 12 Wochen behandelt wurden, zeigten 4 %, 2 %, 6 % bzw. 13 % eine anormale stimulierte Cortisol-Reaktion (Spitzen-Cortisol
Andere Formoterol-Produkte
Während die pharmakodynamische Wirkung über die Stimulation von beta-adrenergen Rezeptoren erfolgt, führt eine übermäßige Aktivierung dieser Rezeptoren häufig zu Skelettmuskelzittern und -krämpfen, Schlaflosigkeit, Tachykardie, Abnahme des Plasmakaliums und Anstieg der Plasmaglukose. Inhaliertes Formoterol kann wie andere beta2-adrenerge Agonisten dosisabhängige kardiovaskuläre Wirkungen und Auswirkungen auf den Blutzucker und/oder das Serumkalium haben [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ]. Für SYMBICORT 160 mg werden diese Wirkungen im Abschnitt Klinische Pharmakologie, Pharmakodynamik, SYMBICORT (12.2) detailliert beschrieben.
Die Anwendung von LABA-Medikamenten kann zu einer Toleranz gegenüber bronchoprotektiven und bronchodilatatorischen Wirkungen führen.
Ein Rebound der bronchialen Hyperreagibilität nach Absetzen einer chronisch langwirksamen Beta-Agonisten-Therapie wurde nicht beobachtet.
Pharmakokinetik
SYMBIKORT
Absorption
Budesonid
Gesunde Probanden
Oral inhaliertes Budesonid wird schnell in die Lunge resorbiert und die Spitzenkonzentration wird typischerweise innerhalb von 20 Minuten erreicht. Nach oraler Gabe von Budesonid wurde die maximale Plasmakonzentration nach etwa 1 bis 2 Stunden erreicht, und die absolute systemische Verfügbarkeit betrug aufgrund des ausgedehnten First-Pass-Metabolismus 6 % bis 13 %. Im Gegensatz dazu wurde das meiste an die Lungen abgegebene Budesonid systemisch absorbiert. Bei gesunden Probanden wurden 34 % der abgemessenen Dosis in der Lunge abgelagert (bestimmt durch Plasmakonzentrationsmethode und unter Verwendung eines Budesonid-haltigen Trockenpulverinhalators) mit einer absoluten systemischen Verfügbarkeit von 39 % der abgemessenen Dosis.
Nach Verabreichung von SYMBICORT 160/4.5, zwei oder vier Inhalationen zweimal täglich über 5 Tage bei gesunden Probanden, stieg die Plasmakonzentration von Budesonid im Allgemeinen proportional zur Dosis an. Der Akkumulationsindex für die Gruppe, die zweimal täglich 2 Inhalationen erhielt, betrug 1,32 für Budesonid.
Asthmapatienten
In einer Einzeldosisstudie wurden Patienten mit mittelschwerem Asthma höhere als die empfohlenen Dosen von SYMBICORT (12 Inhalationen von SYMBICORT 160/4,5) verabreicht. Die maximale Budesonid-Plasmakonzentration von 4,5 nmol/l trat 20 Minuten nach der Einnahme auf. Diese Studie zeigte, dass die systemische Gesamtexposition von Budesonid durch SYMBICORT 4,5 mcg etwa 30 % niedriger war als durch inhaliertes Budesonid über einen Trockenpulverinhalator (DPI) bei derselben verabreichten Dosis. Nach Verabreichung von SYMBICORT betrug die Halbwertszeit der Budesonid-Komponente 4,7 Stunden.
In einer Studie mit wiederholter Gabe wurde die höchste empfohlene Dosis von SYMBICORT (160/4,5, zwei Inhalationen zweimal täglich) Patienten mit mittelschwerem Asthma und gesunden Probanden 1 Woche lang verabreicht. Die maximale Budesonid-Plasmakonzentration von 1,2 nmol/l trat bei Asthmapatienten nach 21 Minuten auf. Die maximale Budesonid-Plasmakonzentration war bei Asthmapatienten um 27 % niedriger als bei gesunden Probanden. Die systemische Gesamtexposition von Budesonid war jedoch mit der bei Asthmapatienten vergleichbar.
Die maximalen Steady-State-Plasmakonzentrationen von Budesonid, das mit DPI verabreicht wurde, lagen bei Erwachsenen mit Asthma im Durchschnitt bei 0,6 bzw. 1,6 nmol/l bei Dosierungen von 180 µg bzw. 360 µg zweimal täglich. Bei Asthmapatienten zeigte Budesonid einen linearen Anstieg der AUC und Cmax mit zunehmender Dosis, sowohl nach einmaliger als auch nach wiederholter Gabe von inhaliertem Budesonid.
COPD-Patienten
In einer Einzeldosisstudie wurden Patienten mit COPD 12 Inhalationen von SYMBICORT 80/4,5 (Gesamtdosis 960/54 Mikrogramm) verabreicht. Eine mittlere Budesonid-Spitzenplasmakonzentration von 3,3 nmol/l trat 30 Minuten nach der Einnahme auf. Die systemische Budesonid-Exposition war zwischen SYMBICORT pMDI und der gleichzeitigen Verabreichung von Budesonid über einen Dosierinhalator und Formoterol über einen Trockenpulverinhalator (960 µg Budesonid und 54 µg Formoterol) vergleichbar. In derselben Studie erhielt eine offene Gruppe von Patienten mit mittelschwerem Asthma ebenfalls die gleiche höhere Dosis von SYMBICORT. Bei Budesonid zeigten COPD-Patienten im Vergleich zu Asthmapatienten eine um 12 % größere AUC und eine um 10 % niedrigere Cmax.
In der 6-monatigen zulassungsrelevanten klinischen Studie zur Lungenfunktion wurden Steady-State-Pharmakokinetikdaten von Budesonid bei einer Untergruppe von COPD-Patienten mit den Behandlungsarmen SYMBICORT 4,5 µg pMDI 160/4,5, SYMBICORT 160 mg pMDI 80/4,5, Budesonid 160 µg, Budesonid erhoben 160 µg und Formoterol 4,5 µg zusammen gegeben, alle verabreicht als 2 Inhalationen zweimal täglich. Die systemische Budesonid-Exposition (AUC und Cmax) stieg proportional mit Dosen von 80 µg bis 160 µg und war im Allgemeinen ähnlich zwischen den 3 Behandlungsgruppen, die dieselbe Budesonid-Dosis erhielten (SYMBICORT 4,5 µg pMDI 160/4,5, Budesonid 160 µg, Budesonid 160 µg und Formoterol 4,5 µg zusammen verabreicht).
Formoterol
Inhaliertes Formoterol wird schnell resorbiert; Spitzenplasmakonzentrationen werden typischerweise zum Zeitpunkt der ersten Plasmaentnahme innerhalb von 5-10 Minuten nach der Verabreichung erreicht. Wie bei vielen Arzneimitteln zur oralen Inhalation ist es wahrscheinlich, dass der Großteil des inhalierten Formoterols geschluckt und dann aus dem Magen-Darm-Trakt resorbiert wird.
Gesunde Probanden
Nach Verabreichung von SYMBICORT (160/4,5, zwei oder vier Inhalationen zweimal täglich) über 5 Tage bei gesunden Probanden stieg die Plasmakonzentration von Formoterol im Allgemeinen proportional zur Dosis an. Der Akkumulationsindex für die Gruppe, die zweimal täglich 2 Inhalationen erhielt, betrug 1,77 für Formoterol.
Asthmapatienten
In einer Einzeldosisstudie wurden Patienten mit mittelschwerem Asthma höhere als die empfohlenen Dosen von SYMBICORT (12 Inhalationen von SYMBICORT 160/4,5) verabreicht. Die maximale Plasmakonzentration von Formoterol von 136 pmol wurde 10 Minuten nach der Einnahme erreicht. Ungefähr 8 % der verabreichten Formoterol-Dosis wurden im Urin als unveränderter Wirkstoff wiedergefunden.
In einer Studie mit wiederholter Gabe wurde die höchste empfohlene Dosis von SYMBICORT (160/4,5, zwei Inhalationen zweimal täglich) Patienten mit mittelschwerem Asthma und gesunden Probanden 1 Woche lang verabreicht. Bei Asthmapatienten trat nach 10 Minuten eine maximale Formoterol-Plasmakonzentration von 28 pmol/l auf. Die maximale Formoterol-Plasmakonzentration war bei Asthmapatienten etwa 42 % niedriger als bei gesunden Probanden. Die systemische Gesamtexposition von Formoterol war jedoch mit der bei Asthmapatienten vergleichbar.
COPD-Patienten
Nach Gabe einer Einzeldosis von 12 Inhalationen von SYMBICORT 80/4,5 wurde 15 Minuten nach der Gabe rasch eine mittlere maximale Formoterol-Plasmakonzentration von 167 pmol/l erreicht. Die Formoterol-Exposition war bei SYMBICORT 160 mg pMDI etwas höher (~16-18 %) im Vergleich zur gleichzeitigen Verabreichung von Budesonid über einen Dosierinhalator und Formoterol über einen Trockenpulverinhalator (Gesamtdosis von Budesonid 960 µg und Formoterol 54 µg). In derselben Studie erhielt eine Open-Label-Gruppe von Patienten mit mittelschwerem Asthma dieselbe Dosis SYMBICORT. COPD-Patienten wiesen im Vergleich zu Asthmapatienten eine um 12-15 % höhere AUC und Cmax für Formoterol auf.
In der 6-monatigen zulassungsrelevanten klinischen Studie zur Lungenfunktion wurden Steady-State-Pharmakokinetikdaten von Formoterol bei einer Untergruppe von COPD-Patienten mit Behandlungsarmen mit SYMBICORT 160 mg pMDI 160/4,5, SYMBICORT 160 mg pMDI 80/4,5, Formoterol 4,5 µg, Budesonid 160 erhoben mcg und Formoterol 4,5 mcg zusammen gegeben, alle verabreicht als 2 Inhalationen zweimal täglich. Die systemische Formoterol-Exposition, nachgewiesen durch die AUC, war etwa 30 % bzw. 16 % höher bei SYMBICORT pMDI im Vergleich zum Behandlungsarm mit Formoterol allein bzw. zur gleichzeitigen Verabreichung einzelner Komponenten von Budesonid bzw. zum Behandlungsarm mit Formoterol.
Verteilung
Budesonid
Das Verteilungsvolumen von Budesonid betrug etwa 3 l/kg. Es war zu 85 % bis 90 % an Plasmaproteine gebunden. Die Proteinbindung war über den Konzentrationsbereich (1–100 nmol/l) konstant, der mit und über den empfohlenen inhalativen Dosen erreicht wurde. Budesonid zeigte wenig oder keine Bindung an Corticosteroid-bindendes Globulin. Budesonid äquilibrierte schnell mit roten Blutkörperchen in einer konzentrationsunabhängigen Weise mit einem Blutplasmaverhältnis von etwa 0,8.
Formoterol
Über den Konzentrationsbereich von 10-500 nmol/l betrug die Plasmaproteinbindung für die RR- und SS-Enantiomere von Formoterol 46 % bzw. 58 %. Die Konzentrationen von Formoterol, die zur Beurteilung der Plasmaproteinbindung verwendet wurden, waren höher als diejenigen, die im Plasma nach Inhalation einer Einzeldosis von 54 Mikrogramm erreicht wurden.
Stoffwechsel
Budesonid
In-vitro-Studien mit humanen Leberhomogenaten haben gezeigt, dass Budesonid schnell und umfassend metabolisiert wird. Zwei Hauptmetaboliten, die durch Cytochrom P450 (CYP)-Isoenzym 3A4 (CYP3A4)-katalysierte Biotransformation gebildet werden, wurden isoliert und als 16α-Hydroxyprednisolon und 6ß-Hydroxybudesonid identifiziert. Die Corticosteroid-Aktivität jedes dieser beiden Metaboliten betrug weniger als 1 % der Aktivität der Ausgangssubstanz. Es wurden keine qualitativen Unterschiede zwischen den Stoffwechselmustern in vitro und in vivo festgestellt. In menschlichen Lungen- und Serumpräparaten wurde eine vernachlässigbare metabolische Inaktivierung beobachtet.
Formoterol
Der primäre Metabolismus von Formoterol erfolgt durch direkte Glucuronidierung und durch O-Demethylierung, gefolgt von Konjugation an inaktive Metaboliten. Sekundäre Stoffwechselwege umfassen Deformylierung und Sulfatkonjugation. CYP2D6 und CYP2C wurden als primär für die Odemethylierung verantwortlich identifiziert.
Beseitigung
Budesonid
Budesonid wurde in Form von Metaboliten im Urin und im Stuhl ausgeschieden. Ungefähr 60 % einer intravenösen radioaktiv markierten Dosis wurden im Urin wiedergefunden.
Im Urin wurde kein unverändertes Budesonid nachgewiesen. Die 22R-Form von Budesonid wurde vorzugsweise von der Leber mit einer systemischen Clearance von 1,4 l/min gegenüber 1,0 l/min für die 22S-Form ausgeschieden. Die terminale Halbwertszeit, 2 bis 3 Stunden, war für beide Epimere gleich und dosisunabhängig.
Formoterol
Die Ausscheidung von Formoterol wurde bei vier gesunden Probanden nach gleichzeitiger oraler und intravenöser Verabreichung von radioaktiv markiertem Formoterol untersucht. In dieser Studie wurden 62 % des radioaktiv markierten Formoterols mit dem Urin ausgeschieden, während 24 % mit den Faeces ausgeschieden wurden.
Besondere Populationen
Geriatrie
Die Pharmakokinetik von SYMBICORT 4,5 µg bei geriatrischen Patienten wurde nicht speziell untersucht.
Pädiatrie
Die Plasmakonzentrationen von Budesonid wurden nach Verabreichung von vier Inhalationen von SYMBICORT 160/4,5 in einer Einzeldosisstudie bei pädiatrischen Patienten mit Asthma im Alter von 6 bis unter 12 Jahren gemessen. Spitzenkonzentrationen von Budesonid von 1,4 nmol/l traten 20 Minuten nach der Einnahme auf. Diese Studie zeigte auch, dass die gesamte systemische Budesonid-Exposition von SYMBICORT 4,5 mcg etwa 30 % niedriger war als die von inhaliertem Budesonid über einen Trockenpulverinhalator, der ebenfalls mit der gleichen verabreichten Dosis bewertet wurde. Die dosisnormalisierte Cmax und AUC0-inf von Budesonid nach Inhalation einer Einzeldosis bei Kindern im Alter von 6 bis unter 12 Jahren waren numerisch niedriger als bei Erwachsenen.
Nach 2 Inhalationen von SYMBICORT 160/4,5 zweimal täglich waren die Cmax und AUC0-6 von Formoterol im Steady State bei Kindern im Alter von 6 bis unter 12 Jahren vergleichbar mit denen, die bei Erwachsenen beobachtet wurden.
Geschlecht/Rasse
Spezifische Studien zur Untersuchung der Auswirkungen von Geschlecht und ethnischer Zugehörigkeit auf die Pharmakokinetik von SYMBICORT wurden nicht durchgeführt. Die populationspharmakokinetische Analyse der Daten von SYMBICORT 4,5 µg weist darauf hin, dass das Geschlecht die Pharmakokinetik von Budesonid und Formoterol nicht beeinflusst. Aufgrund der geringen Anzahl von Nicht-Kaukasiern, die auf PK untersucht wurden, können keine Rückschlüsse auf die Auswirkung der Rasse gezogen werden.
Stillende Mutter
Die Disposition von Budesonid nach Verabreichung durch Inhalation aus einem Trockenpulverinhalator in Dosen von 200 oder 400 Mikrogramm zweimal täglich für mindestens 3 Monate wurde bei acht stillenden Frauen mit Asthma im Zeitraum von 1 bis 6 Monaten nach der Geburt untersucht. Die systemische Budesonid-Exposition bei diesen Frauen scheint mit derjenigen bei nicht-stillenden Frauen mit Asthma aus anderen Studien vergleichbar zu sein. Muttermilch, die über acht Stunden nach der Einnahme gewonnen wurde, ergab, dass die maximale Konzentration von Budesonid für die Tagesgesamtdosen von 400 und 800 Mikrogramm 0,39 bzw. 0,78 nmol/l betrug und innerhalb von 45 Minuten nach der Einnahme auftrat. Die geschätzte orale Tagesdosis von Budesonid aus der Muttermilch an den Säugling beträgt ungefähr 0,007 und 0,014 µg/kg/Tag für die beiden in dieser Studie verwendeten Dosierungsschemata, was ungefähr 0,3 % bis 1 % der von der Mutter inhalierten Dosis entspricht. Budesonidspiegel in Plasmaproben von fünf Säuglingen etwa 90 Minuten nach dem Stillen (und etwa 140 Minuten nach Verabreichung des Arzneimittels an die Mutter) lagen unter quantifizierbaren Spiegeln ( Verwendung in bestimmten Bevölkerungsgruppen ].
Nieren- oder Leberinsuffizienz
Es liegen keine Daten zur spezifischen Anwendung von SYMBICORT bei Patienten mit eingeschränkter Leber- oder Nierenfunktion vor. Eine eingeschränkte Leberfunktion kann die Ausscheidung von Kortikosteroiden beeinträchtigen. Die Pharmakokinetik von Budesonid wurde durch eine beeinträchtigte Leberfunktion beeinflusst, was durch eine doppelte systemische Verfügbarkeit nach oraler Einnahme belegt wurde. Die Pharmakokinetik von intravenösem Budesonid war jedoch bei Patienten mit Zirrhose und bei gesunden Probanden ähnlich. Spezifische Daten zu Formoterol sind nicht verfügbar, aber da Formoterol hauptsächlich über den Leberstoffwechsel ausgeschieden wird, ist bei Patienten mit schwerer Leberfunktionsstörung mit einer erhöhten Exposition zu rechnen.
Arzneimittelwechselwirkungen
Eine Einzeldosis-Crossover-Studie wurde durchgeführt, um die Pharmakokinetik von acht Inhalationen der folgenden Substanzen zu vergleichen: Budesonid, Formoterol und gleichzeitig verabreichtes Budesonid plus Formoterol. Die Ergebnisse der Studie deuteten darauf hin, dass es keine Hinweise auf eine pharmakokinetische Wechselwirkung zwischen den beiden Bestandteilen von SYMBICORT gab.
Inhibitoren von Cytochrom-P450-Enzymen
Ketoconazol
Ketoconazol, ein starker Inhibitor des Isoenzyms 3A4 (CYP3A4) von Cytochrom P450 (CYP), dem wichtigsten Stoffwechselenzym für Kortikosteroide, erhöhte die Plasmaspiegel von oral eingenommenem Budesonid.
Cimetidin
Bei empfohlener Dosierung hatte Cimetidin, ein unspezifischer Inhibitor von CYP-Enzymen, eine leichte, aber klinisch unbedeutende Wirkung auf die Pharmakokinetik von oralem Budesonid.
Spezifische Arzneimittelwechselwirkungsstudien mit Formoterol wurden nicht durchgeführt.
Tiertoxikologie und/oder Pharmakologie
Präklinisch
Studien an Labortieren (Minischweine, Nagetiere und Hunde) haben das Auftreten von Herzrhythmusstörungen und plötzlichem Herztod (mit histologischem Nachweis einer Myokardnekrose) gezeigt, wenn Beta-Agonisten und Methylxanthine gleichzeitig verabreicht wurden. Die klinische Bedeutung dieser Befunde ist nicht bekannt.
Klinische Studien
Asthma
Patienten mit Asthma im Alter von 12 Jahren und älter
In zwei klinischen Studien, in denen SYMBICORT 4,5 µg mit den einzelnen Bestandteilen verglichen wurde, waren die Verbesserungen bei den meisten Wirksamkeitsendpunkten bei SYMBICORT größer als bei der alleinigen Anwendung von entweder Budesonid oder Formoterol. Darüber hinaus zeigte eine klinische Studie ähnliche Ergebnisse zwischen SYMBICORT und der gleichzeitigen Anwendung von Budesonid und Formoterol in entsprechenden Dosen aus separaten Inhalatoren.
Die Sicherheit und Wirksamkeit von SYMBICORT 160 mg wurden in zwei randomisierten, doppelblinden, placebokontrollierten klinischen Studien in den USA mit 1076 Patienten ab 12 Jahren nachgewiesen. Fixe SYMBICORT-Dosierungen von 160/9 µg und 320/9 µg zweimal täglich (jede Dosis verabreicht als 2 Inhalationen der Stärken 80/4,5 bzw. 160/4,5 µg) wurden mit den Monokomponenten (Budesonid und Formoterol) und Placebo verglichen Informationen zur angemessenen Dosierung zur Verfügung stellen, um eine Reihe von Asthma-Schweregraden abzudecken.
Studie 1: Klinische Studie mit SYMBICORT 160/4.5
In dieser 12-wöchigen Studie wurden 596 Patienten im Alter von 12 Jahren und älter untersucht, indem SYMBICORT 160/4,5, die freie Kombination aus Budesonid 160 µg plus Formoterol 4,5 µg in separaten Inhalatoren, Budesonid 160 µg, Formoterol 4,5 µg und Placebo; jeweils als 2 Inhalationen zweimal täglich verabreicht. Die Studie umfasste eine 2-wöchige Einlaufphase mit Budesonid 80 mcg, 2 Inhalationen zweimal täglich. Die meisten Patienten hatten mittelschweres bis schweres Asthma und verwendeten vor Studienbeginn mäßige bis hohe Dosen inhalativer Kortikosteroide. Die Randomisierung wurde nach vorheriger Behandlung mit inhalativen Kortikosteroiden stratifiziert (71,6 % mit mäßig dosiertem und 28,4 % mit hoch dosiertem inhalativem Kortikosteroid). Der mittlere Prozentsatz des vorhergesagten FEV1 zu Studienbeginn betrug 68,1 % und war in allen Behandlungsgruppen ähnlich. Die co-primären Wirksamkeitsendpunkte waren der 12-Stunden-Durchschnitt des Postdosis-FEV1 in Woche 2 und der Prädosis-FEV1, gemittelt über den Verlauf der Studie. Die Studie erforderte auch, dass Patienten, die ein vordefiniertes Asthma-Verschlechterungskriterium erfüllten, aus der Studie ausgeschlossen wurden. Die vordefinierten Asthma-Verschlimmerungskriterien waren eine klinisch relevante Abnahme von FEV1 oder PEF, eine Erhöhung der Albuterol-Notfallanwendung, nächtliches Erwachen aufgrund von Asthma, Notfallmaßnahmen oder Krankenhausaufenthalt aufgrund von Asthma oder die Notwendigkeit einer Asthmamedikation, die laut Protokoll nicht zulässig ist. Für das Kriterium des nächtlichen Erwachens aufgrund von Asthma durften die Patienten nach Ermessen des Prüfarztes in der Studie bleiben, wenn keines der anderen Asthma-verschlechternden Kriterien erfüllt war. Der prozentuale Anteil der Patienten, die aufgrund von oder Erfüllung vordefinierter Kriterien für eine Verschlechterung des Asthmas abbrachen, ist in Tabelle 4 dargestellt.
Die mittlere prozentuale Veränderung des FEV1 gegenüber dem Ausgangswert, gemessen unmittelbar vor der Verabreichung (vor der Dosis) über 12 Wochen, ist in Abbildung 1 dargestellt. Dosis-FEV1-Ergebnisse beim letzten verfügbaren Studienbesuch (Ende der Behandlung, EOT) werden ebenfalls bereitgestellt. Patienten, die SYMBICORT 160/4,5 erhielten, zeigten am Ende der Behandlung signifikant stärkere mittlere Verbesserungen des FEV1 vor der Dosisgabe gegenüber dem Ausgangswert (0,19 l, 9,4 %) im Vergleich zu Budesonid 160 µg (0,10 l, 4,9 %), Formoterol 4,5 µg (-0,12 L, -4,8 %) und Placebo (-0,17 L, -6,9 %).
Abbildung 1: Mittlere prozentuale Veränderung des FEV1 vor der Dosisgabe gegenüber dem Ausgangswert über 12 Wochen (Studie 1)
INFORMATIONEN ZUM PATIENTEN
SYMBIKORT (SIM-bi-kort) (Budesonid 80 µg und Formoterolfumarat-Dihydrat 4,5 µg) Aerosol zur Inhalation
SYMBIKORT (SIM-bi-kort) (Budesonid 160 µg und Formoterolfumarat-Dihydrat 4,5 µg) Aerosol zur Inhalation
Was ist SYMBICORT?
SYMBICORT kombiniert ein inhalatives Kortikosteroid-Arzneimittel (ICS), Budesonid und ein langwirksames Beta2-adrenerge Agonist (LABA)-Arzneimittel, Formoterol.
SYMBICORT 4,5 µg wird nicht zur Linderung plötzlicher Atemprobleme verwendet und ersetzt keinen Notfallinhalator. SYMBICORT wird bei Asthma und COPD wie folgt angewendet:
Verwenden Sie SYMBICORT nicht:
Informieren Sie vor der Anwendung von SYMBICORT Ihren Arzt über alle Ihre Erkrankungen, auch wenn Sie:
Informieren Sie Ihren Arzt über alle Arzneimittel, die Sie einnehmen, einschließlich verschreibungspflichtiger und rezeptfreier Arzneimittel, Vitamine und pflanzlicher Nahrungsergänzungsmittel. SYMBICORT und bestimmte andere Arzneimittel können miteinander interagieren. Dies kann schwerwiegende Nebenwirkungen verursachen. Informieren Sie Ihren Arzt insbesondere, wenn Sie Antimykotika oder Anti-HIV-Arzneimittel einnehmen.
Kennen Sie alle Medikamente, die Sie einnehmen. Führen Sie eine Liste und zeigen Sie sie Ihrem Arzt und Apotheker jedes Mal, wenn Sie ein neues Arzneimittel erhalten.
Wie sollte ich SYMBICORT verwenden?
Siehe Schritt-für-Schritt-Anleitung zur Anwendung von SYMBICORT am Ende dieser Packungsbeilage. Verwenden Sie SYMBICORT 4,5 mcg nicht, es sei denn, Ihr medizinischer Betreuer hat es Ihnen beigebracht und Sie verstehen alles. Wenden Sie sich bei Fragen an Ihren Arzt oder Apotheker.
Welche Nebenwirkungen kann SYMBICORT haben?
SYMBICORT 4,5 mcg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Zu den häufigsten Nebenwirkungen von SYMBICORT gehören:
Menschen mit Asthma:
Menschen mit COPD:
Informieren Sie Ihren Arzt über jede Nebenwirkung, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von SYMBICORT.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
Sie können Nebenwirkungen auch an AstraZeneca unter 1-800-236-9933 melden.
Wie ist SYMBICORT aufzubewahren?
Allgemeine Informationen zur sicheren und wirksamen Anwendung von SYMBICORT.
Arzneimittel werden manchmal zu anderen als den in der Packungsbeilage aufgeführten Zwecken verschrieben. Verwenden Sie SYMBICORT 4,5 mcg nicht für einen Zustand, für den es nicht verschrieben wurde. Geben Sie SYMBICORT nicht an andere Personen weiter, selbst wenn diese die gleichen Symptome wie Sie haben. Es kann ihnen schaden.
Sie können Ihren Arzt oder Apotheker um Informationen zu SYMBICORT 4,5 mcg bitten, die für Angehörige der Gesundheitsberufe bestimmt sind.
Welche Inhaltsstoffe enthält SYMBICORT 4,5 mcg?
Wirkstoffe: mikronisiertes Budesonid und mikronisiertes Formoterolfumarat-Dihydrat
Inaktive Inhaltsstoffe: Hydrofluoralkan (HFA 227), Povidon K25 USP und Polyethylenglykol 1000 NF
Gebrauchsanweisung
SYMBIKORT (SIM-bi-kort)(Budesonid 80 mcg und Formoterolfumarat-Dihydrat 4,5 mcg) Aerosol zur Inhalation
SYMBIKORT (SIM-bi-kort) (Budesonid 160 µg und Formoterolfumarat-Dihydrat 4,5 µg) Aerosol zur Inhalation
Abbildung 1  Aufrechte Position
Aufrechte Position
So verwenden Sie SYMBICORT
Befolgen Sie die nachstehenden Anweisungen zur Verwendung von SYMBICORT. Sie werden das Arzneimittel einatmen (inhalieren). Wenn Sie Fragen haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Vorbereiten Ihres SYMBICORT-Inhalators für die Anwendung
Figur 2 
5. Vorbereiten Ihres SYMBICORT-Inhalators
Bevor Sie SYMBICORT zum ersten Mal verwenden, müssen Sie es vorbereiten. Um SYMBICORT 160 mg zu entlüften, halten Sie es in aufrechter Position. Siehe Abbildung 1. Schütteln Sie den SYMBICORT 4,5-mcg-Inhalator 5 Sekunden lang gut. Halten Sie Ihren SYMBICORT 160 mg-Inhalator von sich weg gerichtet und drücken Sie fest und vollständig auf die Oberseite des Zählers auf dem SYMBICORT 160 mg-Inhalator, um einen Testspray freizusetzen. Schütteln Sie es dann erneut für 5 Sekunden und geben Sie einen zweiten Testsprühstoß ab. Ihr SYMBICORT 160 mg-Inhalator ist jetzt vorbereitet und gebrauchsfertig. Nachdem Sie den SYMBICORT-Inhalator zum ersten Mal gefüllt haben, zeigt der Zähler entweder 120 oder 60 an, je nachdem, welche Größe Sie erhalten haben.
Wenn Sie Ihren SYMBICORT-Inhalator länger als 7 Tage nicht verwenden oder wenn Sie ihn fallen lassen, müssen Sie ihn erneut entlüften.
Möglichkeiten, den SYMBICORT 160-mg-Inhalator für die Anwendung zu halten
Figur 3 
ODER
Figur 4 
Anwendung Ihres SYMBICORT-Inhalators
6. Schütteln Sie Ihren SYMBICORT 160 mg Inhalator 5 Sekunden lang gut. Entfernen Sie die Mundstückabdeckung. Überprüfen Sie das Mundstück auf Fremdkörper.
7. Atmen Sie vollständig aus (ausatmen). Halten Sie den SYMBICORT-Inhalator an Ihren Mund. Setzen Sie das weiße Mundstück vollständig in Ihren Mund ein und schließen Sie Ihre Lippen darum. Stellen Sie sicher, dass der SYMBICORT 4,5-mcg-Inhalator aufrecht steht und dass die Öffnung des Mundstücks in Richtung Ihres Rachens zeigt (siehe Abbildung 5).
Abbildung 5 
8. Atmen Sie tief und langsam durch den Mund ein (einatmen). Drücken Sie fest und vollständig auf die Oberseite des Zählers am SYMBICORT-Inhalator, um das Arzneimittel freizusetzen (siehe Abbildungen 3 und 4).
9. Atmen Sie weiter ein (einatmen) und halten Sie den Atem etwa 10 Sekunden oder so lange an, wie es angenehm ist. Bevor Sie ausatmen (ausatmen), lösen Sie Ihren Finger von der Oberseite des Zählers. Halten Sie den SYMBICORT-Inhalator aufrecht und nehmen Sie ihn aus dem Mund.
10. Schütteln Sie den SYMBICORT-Inhalator erneut 5 Sekunden lang und wiederholen Sie die Schritte 7 bis 9.
Nach der Anwendung Ihres SYMBICORT-Inhalators
11. Schließen Sie nach Gebrauch die Mundstückabdeckung, indem Sie sie drücken, bis sie einrastet.
12. Nachdem Sie die Einnahme von SYMBICORT (2 Sprühstöße) beendet haben, spülen Sie Ihren Mund mit Wasser aus. Spucke das Wasser aus. Schlucken Sie es nicht.
Ablesen des Zählers
ZÄHLER 
ZÄHLER 
So reinigen Sie Ihren SYMBICORT-Inhalator
Reinigen Sie das weiße Mundstück Ihres SYMBICORT 4,5-µg-Inhalators alle 7 Tage. So reinigen Sie das Mundstück:
Diese Patienteninformation und Gebrauchsanweisung wurde von der US Food and Drug Administration genehmigt.
Was ist Symmetrel und wie wird es angewendet?
Symmetrel 100 mg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Influenza A und der Parkinson-Krankheit. Symmetrel kann allein oder mit anderen Medikamenten verwendet werden.
Symmetrel gehört zu einer Klasse von Medikamenten namens Antiparkinson-Mittel, Dopamin-Agonisten; Virostatika, Grippe.
Es ist nicht bekannt, ob Symmetrel 100 mg bei Kindern unter 1 Jahr sicher und wirksam ist.
Welche Nebenwirkungen kann Symmetrel 100 mg haben?
Symmetrel kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Symmetrel gehören:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Symmetrel. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
SYMMETREL (Amantadinhydrochlorid, USP) wird generisch als Amantadinhydrochlorid und chemisch als 1-Adamantanaminhydrochlorid bezeichnet.
Amantadinhydrochlorid ist ein stabiles weißes oder fast weißes kristallines Pulver, das in Wasser frei löslich und in Alkohol und Chloroform löslich ist.
Amantadinhydrochlorid hat pharmakologische Wirkungen sowohl als Anti-Parkinson- als auch als antivirales Medikament.
SYMMETREL (Amantadinhydrochlorid) ist in Tabletten und Sirup erhältlich.
Jede zur oralen Verabreichung bestimmte Tablette enthält 100 mg Amantadinhydrochlorid und hat die folgenden inaktiven Bestandteile: Hydroxypropylmethylcellulose, Magnesiumstearat, mikrokristalline Cellulose, Natriumstärkeglykolat, FD&C Yellow Nr. 6.
SYMMETREL 100 mg Sirup enthält 50 mg Amantadinhydrochlorid pro 5 ml und hat die folgenden inaktiven Inhaltsstoffe: künstliches Himbeeraroma, Zitronensäure, Methylparaben, Propylparaben und Sorbitlösung.
INDIKATIONEN
Behandlung der Parkinson-Krankheit
Vorbeugung und Behandlung von Anzeichen und Symptomen einer Infektion, die durch verschiedene Stämme des Influenza-A-Virus verursacht werden
Amantadin ist zur Behandlung einer aktiven Influenza-A-Infektion indiziert, wenn es innerhalb von 48 Stunden nach Auftreten der Symptome verabreicht wird.
Bei pädiatrischen Patienten ist Symmetrel nur bei Kindern ab 5 Jahren indiziert.
Bei der Anwendung von Symmetrel, sei es als Einzelperson oder in Patientengruppen, ist es wichtig, dass die Behandlung unter ärztlicher Aufsicht erfolgt.
DOSIERUNG UND ANWENDUNG
Dosierungsschema
Allgemeine Zielgruppe
Parkinson-Krankheit
Anfänglich 100 mg pro Tag, Steigerung nach einer Woche auf 200 mg pro Tag in 2 aufgeteilten Dosen. Die Dosis kann je nach Anzeichen und Symptomen titriert werden. Mengen von mehr als 200 mg täglich (bis zu einer Höchstdosis von 300 mg täglich in aufgeteilten Dosen) können eine gewisse zusätzliche Linderung bewirken, können aber auch mit einer zunehmenden Toxizität einhergehen. In diesen Fällen sollte die Dosis schrittweise in Abständen von mindestens 1 Woche erhöht werden. Amantadin wirkt innerhalb weniger Tage, scheint jedoch häufig innerhalb weniger Monate nach kontinuierlicher Behandlung einen Teil seiner Wirksamkeit zu verlieren.
Die Wirksamkeit von Amantadin kann durch vorübergehendes Absetzen, das die Aktivität wiederherzustellen scheint, verlängert werden.
Die Behandlung mit Symmetrel 100 mg muss schrittweise reduziert werden, da ein abruptes Absetzen das Parkinson-Syndrom verschlimmern kann, unabhängig davon, wie der Patient auf die Therapie anspricht (siehe Abschnitt WARNUNGEN UND VORSICHTSMASSNAHMEN ).
Symmetrel sollte zu den Mahlzeiten eingenommen werden, vorzugsweise morgens und mittags.
Kombinierte Behandlung: Andere Antiparkinson-Medikamente, mit denen der Patient bereits behandelt wird, sollten während der ersten Phase der Behandlung mit Symmetrel fortgesetzt werden. In vielen Fällen ist es dann möglich, die Dosierung des anderen Medikaments schrittweise zu reduzieren, ohne das Ansprechen auf die Behandlung zu beeinträchtigen. Treten jedoch vermehrt Nebenwirkungen auf, sollte die Dosierung schneller reduziert werden. Bei Patienten, die bereits hohe Dosen von Anticholinergika oder L-Dopa erhalten, sollte die initiale niedrig dosierte Phase der Behandlung mit Symmetrel 100 mg auf 15 Tage verlängert werden.
Arzneimittelinduzierter Parkinsonismus
Wenn die Behandlung von arzneimittelinduziertem Parkinsonismus durch Dosisreduktion des verursachenden Arzneimittels nicht praktikabel ist, kann Symmetrel mit 100 mg zweimal täglich begonnen werden. Die Behandlung mit Symmetrel 100 mg kann reduziert werden, wenn die extrapyramidalen Symptome für eine gewisse Zeit unter Kontrolle sind.
Typ-A-Virus-Influenza – Vorbeugung und Behandlung
Erwachsene (im Alter von 19-65 Jahren): 200 mg pro Tag in 2 aufgeteilten Dosen. .
Es wurde über eine wirksame Vorbeugung und Behandlung von Influenza A mit einer Dosis von 100 mg täglich berichtet (bei Unverträglichkeit gegenüber 200 mg pro Tag könnte eine Dosis von 100 mg täglich verwendet werden).
Prävention: Zur Prophylaxe sollte dieses Regime in Erwartung eines Kontakts begonnen und für die Dauer des Influenza-A-Ausbruchs fortgesetzt werden, normalerweise für etwa 6 Wochen. Bei Anwendung mit einem inaktivierten Influenza-A-Impfstoff sollte Amantadin für 2 bis 3 Wochen nach Verabreichung des Impfstoffs fortgesetzt werden.
Behandlung: Es ist ratsam, die Behandlung der Influenza so früh wie möglich zu beginnen und 4 bis 5 Tage fortzusetzen. Wenn mit der Behandlung mit Amantadin innerhalb von 48 Stunden nach Auftreten der Symptome begonnen wird, wird die Dauer von Fieber und anderen Wirkungen um 1 Tag reduziert und die Entzündungsreaktion des kleinen Bronchialbaums, die normalerweise mit einer Influenza einhergeht, klingt schneller ab.
Besondere Populationen
Pädiatrische Patienten (unter 18 Jahren)
Typ-A-Virus-Grippe – Vorbeugung und Behandlung
Geriatrische Patienten (65 Jahre oder älter)
Die Plasmakonzentration von Amantadin wird durch die Nierenfunktion beeinflusst. Bei älteren Menschen ist die Eliminationshalbwertszeit tendenziell länger und die renale Clearance geringer als bei jüngeren Personen. Daher wird bei älteren Patienten ohne Nierenerkrankung eine Dosis von nicht mehr als 100 mg täglich empfohlen. Wenn der Patient eine eingeschränkte Nierenfunktion hat, sollte das Dosierungsintervall angepasst werden (siehe Abschnitt DOSIERUNG UND ANWENDUNG - Nierenfunktionsstörung ).
Nierenfunktionsstörung
Bei Patienten mit eingeschränkter Nierenfunktion und unter Hämodialyse ist die Eliminationshalbwertszeit von Amantadin erheblich verlängert, was zu erhöhten Plasmakonzentrationen führt. Bei diesen Patienten ist eine sorgfältige Anpassung der Dosis von Symmetrel 100 mg durch Verlängerung des Dosierungsintervalls entsprechend der Kreatinin-Clearance (siehe Tabelle 1) erforderlich.
Patienten mit eingeschränkter Nierenfunktion, die wegen einer Influenza-A-Infektion behandelt werden, sollte am ersten Tag eine Aufsättigungsdosis von 200 mg Symmetrel verabreicht werden. Bei Patienten mit eingeschränkter Nierenfunktion, die eine Behandlung mit Symmetrel 100 mg gegen die Parkinson-Krankheit beginnen, sollte am ersten Tag eine Aufsättigungsdosis von 100 mg/Tag angewendet werden.
Dosis danach: 100 mg in den unten gezeigten Intervallen
Hämodialysepatienten sollte ab dem 8. Tag einmal alle 7 Tage (dh einmal wöchentlich) eine Erhaltungsdosis von 100 mg gegeben werden.
Idealerweise sollten die Plasmakonzentrationen von Amantadin überwacht werden. Eine sorgfältige Überwachung des Patienten wird empfohlen (siehe Abschnitt KLINISCHE PHARMAKOLOGIE/Pharmakokinetik/Besondere Patientengruppen ).
Art der Verabreichung
Die Kapseln sollten oral mit Nahrung eingenommen werden, um Magenreizungen zu vermeiden.
WIE GELIEFERT
Lagerung und Handhabung
Siehe Faltschachtel.
Symmetrel 100 mg darf nach dem auf der Packung mit „EXP“ gekennzeichneten Datum nicht mehr verwendet werden.
Hersteller: Novartis Pharma AG, Basel, Schweiz. Überarbeitet: April 2019.
NEBENWIRKUNGEN
Die Nebenwirkungen von Amantadin sind oft milder und vorübergehender Natur. Sie treten normalerweise innerhalb der ersten 2-4 Tage der Behandlung auf und verschwinden umgehend innerhalb von 24-48 Stunden nach Absetzen von Amantadin.
Ein direkter Zusammenhang zwischen Dosis und Auftreten von Nebenwirkungen wurde nicht nachgewiesen; es scheint jedoch eine Tendenz zu häufigeren unerwünschten Arzneimittelwirkungen (insbesondere mit Auswirkungen auf das Zentralnervensystem) bei steigenden Dosen zu geben.
Nebenwirkungen aus klinischen Studien, Spontanmeldungen und Literaturfällen (Tabelle 2) sind nach MedDRA-Systemorganklassen aufgelistet. Innerhalb jeder Systemorganklasse werden die Nebenwirkungen nach Häufigkeit geordnet, wobei die häufigsten Nebenwirkungen zuerst aufgeführt werden. Innerhalb jeder Häufigkeitsgruppe werden die unerwünschten Arzneimittelwirkungen nach abnehmendem Schweregrad aufgeführt. Darüber hinaus basiert die entsprechende Häufigkeitskategorie für jede Nebenwirkung auf der folgenden Konvention (CIOMS III): sehr häufig (≥1/10); häufig (≥1/100 bis
Zusätzliche Nebenwirkungen aus Spontanmeldungen und Literaturfällen (Häufigkeit nicht bekannt)
Störungen der Impulskontrolle
Pathologisches Spielen, gesteigerte Libido, Hypersexualität, zwanghaftes Geldausgeben oder Kaufen, Essattacken und zwanghaftes Essen können bei Patienten auftreten, die mit Dopaminagonisten, einschließlich Symmetrel, behandelt werden (siehe Abschnitt WARNUNGEN UND VORSICHTSMASSNAHMEN ).
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Beobachtete Wechselwirkungen, die dazu führen, dass die gleichzeitige Anwendung nicht empfohlen wird
Die gleichzeitige Anwendung von Amantadin mit einer Fixdosiskombination aus Hydrochlorothiazid und Triamteren kann die systemische Clearance des Arzneimittels verringern, was zu erhöhten Plasmakonzentrationen und toxischen Wirkungen (Verwirrtheit, Halluzinationen, Ataxie, Myoklonus) führen kann.
In Einzelfällen wurde bei Patienten, die Amantadin und gleichzeitig Antipsychotika oder Levodopa erhielten, über psychotische Dekompensation berichtet. Die gleichzeitige Anwendung von Amantadin und Anticholinergika oder Levodopa kann Verwirrtheit, Halluzinationen, Albträume, gastrointestinale Störungen oder andere Atropin-ähnliche Nebenwirkungen verstärken (siehe Abschnitt ÜBERDOSIS ).
Erwartete zu berücksichtigende Wechselwirkungen
Medikamente, die auf das zentrale Nervensystem einwirken
Die gleichzeitige Verabreichung von Amantadin und Arzneimitteln oder Substanzen (z. B. Alkohol), die auf das zentrale Nervensystem wirken, kann zu einer additiven ZNS-Toxizität führen. Genaue Beobachtung wird empfohlen (siehe Abschnitt ÜBERDOSIS ).
WARNUNGEN
Eingeschlossen als Teil der "VORSICHTSMASSNAHMEN" Abschnitt
VORSICHTSMASSNAHMEN
Es wurde berichtet, dass Patienten mit vorbestehenden Anfallsleiden während der Amantadin-Therapie häufiger generalisierte Anfälle entwickeln. Eine Dosisreduktion kann dieses Risiko minimieren. Diese Patienten sollten engmaschig überwacht werden.
Bei Patienten mit psychiatrischen Grunderkrankungen kann es zu einer Zunahme von Halluzinationen, Verwirrtheit und Albträumen kommen.
Aufgrund der Möglichkeit schwerwiegender Nebenwirkungen ist bei der Verschreibung von Symmetrel 100 mg an Patienten, die mit Arzneimitteln behandelt werden, die ZNS-Wirkungen haben, oder bei denen die Risiken den Nutzen der Behandlung überwiegen, Vorsicht geboten. Während der Behandlung mit Amantadin wurde über eine kleine Anzahl von Suizidversuchen berichtet, von denen einige tödlich verliefen. Da einige Patienten auch versucht haben, sich das Leben zu nehmen, indem sie eine Überdosis Amantadin angewendet haben, sollten Rezepte für die kleinste Menge ausgestellt werden, die mit einem guten Patientenmanagement vereinbar ist.
Während der Behandlung mit Symmetrel können periphere Ödeme auftreten, die wahrscheinlich auf lokale Gefäßstörungen zurückzuführen sind. Dies sollte bei Patienten mit Herzinsuffizienz in der Anamnese berücksichtigt werden.
Besondere Vorsicht ist bei Patienten geboten, die an wiederkehrenden Ekzemen, Magengeschwüren oder Herz-Kreislauf-Erkrankungen leiden oder in der Vorgeschichte aufgetreten sind.
Symmetrel sollte bei Patienten mit Leber- oder Nierenerkrankungen mit Vorsicht angewendet werden. Bei eingeschränkter Nierenfunktion sollte die Dosierung entsprechend der Kreatinin-Clearance des einzelnen Patienten angepasst und idealerweise die Amantadin-Plasmakonzentrationen überwacht werden. Da nur geringe Mengen Amantadin von Patienten ausgeschieden werden, die sich einer Hämodialyse wegen Niereninsuffizienz unterziehen, sollte bei diesen Patienten die Dosierung sorgfältig angepasst werden, um Nebenwirkungen zu vermeiden (siehe Abschnitt DOSIERUNG UND ANWENDUNG und Abschnitt ÜBERDOSIS ).
Bei Kindern wurde Hypothermie beobachtet. Bei der Verschreibung von Symmetrel 100 mg an Kinder zur Vorbeugung und Behandlung einer Influenza-Typ-A-Virus-Erkrankung ist Vorsicht geboten (siehe auch Abschnitt DOSIERUNG UND ANWENDUNG ).
Da Amantadin anticholinerge Wirkungen hat, sollte es Patienten mit unbehandeltem Engwinkelglaukom nicht verabreicht werden.
Bei verschwommenem Sehen oder anderen Sehproblemen sollte zum Ausschluss eines Hornhautödems ein Augenarzt aufgesucht werden. Falls ein Hornhautödem diagnostiziert wird, sollte die Behandlung mit Amantadin abgebrochen werden.
Abbruch der Behandlung
Ein abruptes Absetzen von Amantadin kann zu einer Verschlechterung der Symptome der Parkinson-Krankheit oder zu Symptomen führen, die dem malignen neuroleptischen Syndrom (MNS), Katatonie sowie zu kognitiven Manifestationen (z. B. Verwirrtheit, Orientierungslosigkeit, Verschlechterung des Geisteszustands, Delirium) ähneln. Es gibt vereinzelte Berichte über einen möglichen Zusammenhang zwischen dem Auftreten oder der Verschlechterung eines NMS oder einer durch Neuroleptika induzierten Katatonie und dem Absetzen von Amantadin bei Patienten, die gleichzeitig Neuroleptika einnehmen. Die Behandlung mit Amantadin sollte daher nicht abrupt abgebrochen werden.
Widerstand
Eine Resistenz gegen Amantadin und Rimantadin wird leicht durch serielle Passage von Influenzavirusstämmen in vitro oder in vivo in Gegenwart des Arzneimittels erreicht. Influenza-A-Viren (kreuz-)resistent gegen Amantadin und Rimantadin können auftreten, wenn diese Arzneimittel zur Behandlung von Influenza-Infektionen angewendet werden. Scheinbare Übertragung arzneimittelresistenter Viren könnte der Grund für das Versagen von Prophylaxe und Behandlung bei Haushaltskontakten und Pflegeheimpatienten gewesen sein. Bisher gibt es jedoch keine Hinweise darauf, dass das resistente Virus eine Krankheit hervorruft, die sich in irgendeiner Weise von der empfindlicher Viren unterscheidet.
Störungen der Impulskontrolle
Die Patienten sollten regelmäßig auf die Entwicklung von Impulskontrollstörungen überwacht werden. Patienten und Betreuer sollten darauf aufmerksam gemacht werden, dass bei Patienten, die mit Dopaminagonisten, einschließlich Symmetrel, behandelt werden, Verhaltenssymptome von Impulskontrollstörungen auftreten können, darunter pathologisches Spielen, gesteigerte Libido, Hypersexualität, zwanghaftes Geldausgeben oder Kaufen, Essattacken und Esszwang. Wenn solche Symptome auftreten, sollte eine Dosisreduktion/ein ausschleichendes Absetzen in Betracht gezogen werden.
Fahren und Bedienen von Maschinen
Patienten, die Symmetrel 100 mg erhalten, sollten darauf hingewiesen werden, dass Schwindel, verschwommenes Sehen und andere zentralnervöse Symptome auftreten (siehe Abschnitt NEBENWIRKUNGEN ) auftreten und die Reaktion des Patienten beeinträchtigen, in diesem Fall sollten sie kein Fahrzeug führen oder Maschinen bedienen.
Schwangerschaft, Stillzeit, Weibchen und Männchen mit reproduktivem Potenzial
Schwangerschaft
Zusammenfassung der Risiken
Symmetrel 100 mg ist während der Schwangerschaft kontraindiziert. Amantadin-bedingte Komplikationen während der Schwangerschaft wurden berichtet
Tierdaten
Studien zur Reproduktionstoxizität wurden an Ratten und Kaninchen durchgeführt. Bei Ratten erwiesen sich orale Dosen von 50 und 100 mg/kg als teratogen
Stillzeit
Zusammenfassung der Risiken
Amantadin geht in die Muttermilch über. Unerwünschte Arzneimittelwirkungen wurden bei gestillten Säuglingen berichtet. Stillende Mütter sollten Symmetrel nicht einnehmen.
Weibchen und Männchen mit reproduktivem Potenzial
Frauen im gebärfähigen Alter müssen während der Behandlung und für 5 Tage nach ihrer letzten Amantadin-Dosis eine wirksame Verhütungsmethode anwenden (Methoden, die zu einer Schwangerschaftsrate von weniger als 1 % führen).
Unfruchtbarkeit
Symmetrel in einer Dosis von 32 mg/kg/Tag (entspricht der empfohlenen Höchstdosis beim Menschen auf mg/m2-Basis), die sowohl männlichen als auch weiblichen Ratten verabreicht wurde, beeinträchtigte die Fertilität (siehe Abschnitt Nichtklinische Toxikologie ).
ÜBERDOSIS
Eine Überdosierung (akute Überdosierung mit einem Vielfachen der maximal empfohlenen Dosis oder Überexposition aufgrund hoher Dosierungen bei älteren und/oder nierengeschädigten Patienten) mit Symmetrel kann tödlich enden (siehe Abschnitt WARNUNGEN UND VORSICHTSMASSNAHMEN ).
Anzeichen und Symptome
Neuromuskuläre Störungen und Symptome einer akuten Psychose sind hervorstechende Merkmale einer akuten Vergiftung mit Amantadin.
Zentrales Nervensystem
Hyperreflexie, motorische Unruhe, Krämpfe, extrapyramidale Zeichen (Torsionskrämpfe, dystonische Körperhaltung), erweiterte Pupillen, Dysphagie, Verwirrtheit, Orientierungslosigkeit, Delirium, visuelle Halluzinationen, Myoklonus, Aggression/Feindseligkeit, Bewusstseinstrübung und Koma.
Atmungssystem
Hyperventilation, Lungenödem, Atemnot, einschließlich Atemnotsyndrom bei Erwachsenen.
Herz-Kreislauf-System
Herzstillstand und plötzlicher Herztod wurden berichtet. Sinustachykardie, Arrhythmie, Bluthochdruck.
Magen-Darm-System
Übelkeit, Erbrechen, Mundtrockenheit.
Nierenfunktion
Harnverhalt, Nierenfunktionsstörung, einschließlich Anstieg des BUN und verminderte Kreatinin-Clearance.
Überdosierung durch kombinierte medikamentöse Behandlung
Die peripheren und zentralen Nebenwirkungen von Anticholinergika werden durch die gleichzeitige Anwendung von Amantadin verstärkt, und es können akute psychotische Reaktionen auftreten, die mit denen einer Atropinvergiftung identisch sein können, wenn hohe Dosen von Anticholinergika angewendet werden. Bei gleichzeitiger Einnahme von Alkohol oder zentralnervösen Stimulanzien können die Anzeichen und Symptome einer akuten Vergiftung mit Amantadin verstärkt und/oder verändert werden.
Management
Es gibt kein spezifisches Gegenmittel.
Entfernung und/oder Inaktivierung von Vergiftungsmitteln: Herbeiführung von Erbrechen und/oder Magenaspiration und Spülung, wenn der Patient bei Bewusstsein ist, Aktivkohle, Kochsalzlösung, falls angemessen. Da Amantadin weitgehend unverändert im Urin ausgeschieden wird, kann es durch Aufrechterhaltung der renalen Ausscheidungsfunktion, reichliche Diurese und gegebenenfalls forcierte Diurese wirksam aus dem Blutkreislauf entfernt werden. Eine Ansäuerung des Urins begünstigt die Ausscheidung von Amantadin im Urin. Hämodialyse entfernt keine signifikanten Mengen von Symmetrel; bei Patienten mit Niereninsuffizienz wurden nach einer oralen Einzeldosis von 300 mg durch 4-stündige Hämodialyse 7 bis 15 mg entfernt.
Überwachung von Blutdruck, Herzfrequenz, EKG, Atmung, Körpertemperatur und gegebenenfalls Behandlung von Hypotonie und Herzrhythmusstörungen. Bei der Verabreichung von adrenergen Substanzen bei Herzrhythmusstörungen und Hypotonie ist Vorsicht geboten, da sich der klinische Zustand aufgrund der arrhythmogenen Natur der adrenergen Arzneimittel verschlechtern kann.
Krämpfe und übermäßige motorische Unruhe: Antikonvulsiva wie Diazepam iv, Paraldehyd im oder per rektum oder Phenobarbital im verabreichen
Akute psychotische Symptome, Delirium, dystonische Körperhaltung, myoklonische Manifestationen: Physostigmin als langsame iv-Infusion (1-mg-Dosen bei Erwachsenen, 0,5-mg-Dosen bei Kindern) in wiederholter Verabreichung entsprechend dem anfänglichen Ansprechen und dem späteren Bedarf wurde berichtet.
Harnretention: Die Blase sollte katheterisiert werden; ein Verweilkatheter kann für die erforderliche Zeit an Ort und Stelle belassen werden.
KONTRAINDIKATIONEN
KLINISCHE PHARMAKOLOGIE
Pharmakotherapeutische Gruppe
Antiparkinsonmittel und antiinfluenzales Virostatikum.
Wirkmechanismus (MOA)/ Pharmakodynamik (PD)
Als Antiparkinsonmittel
Es wird angenommen, dass Amantadin wirkt, indem es die Freisetzung von Dopamin aus zentralen Neuronen verstärkt und seine Wiederaufnahme in synaptische Vesikel verzögert.
Es kann auch eine gewisse anticholinerge Aktivität ausüben.
Wenn es entweder allein oder in Kombination mit anderen Arzneimitteln verabreicht wird, verbessert Amantadin sowohl die Kardinalzeichen und -symptome von Parkinsonismus als auch die funktionelle Kapazität.
Die Wirkung setzt in der Regel 2 bis 5 Tage nach Behandlungsbeginn ein. Es übt eine positive Wirkung aus, insbesondere auf Akinesie, Starrheit und Tremor.
Als Anti-Influenzal Virostatikum
Amantadin hemmt in geringen Konzentrationen spezifisch die Replikation von Influenza-A-Viren. Unter Verwendung eines empfindlichen Plaque-Reduktionsassays werden menschliche Influenza-A-Viren, einschließlich der Subtypen H1N1, H2N2, H3N2, durch 0,4 Mikrogramm/ml oder weniger Amantadin gehemmt. Der genaue Mechanismus der antiviralen Wirkung von Amantadin ist noch nicht vollständig aufgeklärt. Amantadin blockiert die Ionenkanalaktivität des viralen M2-Proteins im Influenzavirus durch allosterische Hemmung, wodurch eine virale Enthüllung nicht stattfinden kann. Dies hemmt schließlich die virale Replikation. Bei bestimmten Vogelgrippeviren wurden Auswirkungen auf späte Replikationsschritte mit beeinträchtigtem Virusaufbau gefunden.
Pharmakokinetik (PK)
Absorption
Amantadin wird langsam, aber fast vollständig resorbiert. Maximale Plasmakonzentrationen von etwa 250 ng/ml und 500 ng/ml werden innerhalb von 3-4 Stunden nach oraler Einzelgabe von 100 mg bzw. 200 mg Amantadin erreicht.
Nach wiederholter Gabe von 200 mg täglich pendelt sich die Steady-State-Plasmakonzentration innerhalb von 3 Tagen auf 300 ng/ml ein.
Verteilung
In vitro werden 67 % von Amantadin an Plasmaproteine gebunden. Eine beträchtliche Menge Amantadin ist an rote Blutkörperchen gebunden. Die Erythrozyten-Amantadin-Konzentration bei normalen gesunden Probanden beträgt das 2,66-fache der Plasmakonzentration.
Das scheinbare Verteilungsvolumen (VD) des Arzneimittels beträgt 5-10 l/kg, was auf eine ausgedehnte Gewebebindung hindeutet. Sie nimmt mit steigender Dosis ab. Die Konzentration von Amantadin in Lunge, Herz, Niere, Leber und Milz ist höher als im Blut.
Das Medikament reichert sich nach mehreren Stunden im Nasensekret an.
Amantadin passiert die Blut-Hirn-Schranke; es ist jedoch nicht möglich, dieses Ereignis zu quantifizieren.
Biotransformation / Stoffwechsel
Amantadin wird in geringem Umfang metabolisiert und es wurden 8 Metaboliten des Arzneimittels identifiziert. Der Hauptmetabolit, der N-acetylierte Metabolit, macht 5-15 % der verabreichten Dosis aus. Die pharmakologische Wirksamkeit oder Toxizität von Metaboliten ist nicht bekannt.
Obwohl der Einfluss des individuellen Acetyliererstatus auf den Metabolismus des Medikaments nicht umfassend untersucht wurde, weisen Studien darauf hin, dass es keine Korrelation zwischen dem NAT1- und NAT2-Acetylierer-Phänotyp und der Amantadin-Acetylierung gibt.
Beseitigung
Amantadin wird bei gesunden jungen Erwachsenen mit einer mittleren Eliminationshalbwertszeit aus dem Plasma von 15 Stunden (10-31 Stunden) ausgeschieden.
Die Gesamtplasmaclearance entspricht etwa der renalen Clearance (250 ml/min). Die renale Amantadin-Clearance ist viel höher als die Kreatinin-Clearance, was auf eine renale tubuläre Sekretion hindeutet.
Eine Einzeldosis Amantadin wird über 72 Stunden wie folgt ausgeschieden: 65-85 % unverändert, 5-15 % als Acetylmetabolit im Urin und 1 % im Stuhl. Nach 4-5 Tagen erscheinen 90 % der Dosis unverändert im Urin. Der pH-Wert des Urins hat einen erheblichen Einfluss auf die Ausscheidungsrate, und ein Anstieg des Urin-pH-Werts kann zu einer erheblichen Verringerung der Ausscheidungsrate von Amantadin führen.
Dosisproportionalität
Amantadin zeigt eine dosisproportionale Pharmakokinetik über einen Dosisbereich von 100 bis 200 mg.
Besondere Populationen
Geschlechtseffekt
Einige wenige Studien weisen auf die Möglichkeit einer höheren renalen Clearance von Amantadin bei Männern als bei Frauen hin.
Geriatrische Patienten
Verglichen mit Daten gesunder junger Erwachsener ist die Halbwertszeit verdoppelt und die renale Clearance verringert. Das renale/Kreatinin-Clearance-Verhältnis ist bei älteren Patienten niedriger als bei jungen Menschen. Im Allgemeinen ist die tubuläre Sekretion bei älteren Menschen stärker reduziert als die glomeruläre Filtration. Bei älteren Patienten mit eingeschränkter Nierenfunktion erhöht die wiederholte Gabe von 100 mg täglich über 14 Tage die Plasmakonzentration in den toxischen Bereich.
Nierenfunktionsstörung
Da Amantadin hauptsächlich über die Nieren ausgeschieden wird, kann es bei Patienten mit eingeschränkter Nierenfunktion akkumulieren. Eine Kreatinin-Clearance von weniger als 40 ml/[min. 1,73 m2] verursacht eine 3- bis 5-fache Erhöhung der Halbwertszeit und eine 5-fache Verringerung der Gesamt- und renalen Clearance. Auch bei Nierenversagen dominiert die renale Ausscheidung.
Ältere Patienten oder Patienten mit Niereninsuffizienz sollten entsprechend der individuellen Kreatinin-Clearance eine entsprechend reduzierte Dosis erhalten. Die angestrebte Amantadin-Plasmakonzentration sollte ein Maximum von 300 Nanogramm/ml nicht überschreiten.
Hämodialyse
Wenig Amantadin wird durch Hämodialyse entfernt; diese Ineffizienz kann mit seiner ausgedehnten Gewebebindung zusammenhängen. Weniger als 5 % einer Dosis werden nach 4-stündiger Hämodialyse ausgeschieden. Die mittlere Halbwertszeit erreicht 24 Dialyse - Stunden.
Leberfunktionsstörung
Die Auswirkung einer Leberfunktionsstörung auf die Pharmakokinetik von Amantadin ist nicht bekannt. Der größte Teil der verabreichten Amantadin-Dosis wird unverändert im Urin ausgeschieden und nur ein kleiner Teil des Arzneimittels wird in der Leber verstoffwechselt (vgl Biotransformation/Stoffwechsel Oben) .
Food-Effekt
Nahrung hat keinen signifikanten Einfluss auf die Pharmakokinetik von Amantadin. Nach Einnahme von Symmetrel zusammen mit Nahrung kann eine leichte Verzögerung beim Einsetzen der Resorption beobachtet werden.
Ethnische Sensibilität
Obwohl der Einfluss von ethnischer Sensibilität und Rasse auf die Pharmakokinetik von Amantadin nicht umfassend untersucht wurde, ist nicht bekannt, dass die Disposition von Amantadin durch genetische Faktoren bestimmt wird (siehe Biotransformation/Stoffwechsel Oben).
Klinische Studien
Symmetrel ist ein etabliertes Produkt. Es wurden keine neueren klinischen Studien durchgeführt.
INFORMATIONEN ZUM PATIENTEN
Gebrauchsanweisung und Handhabung
Hinweis: Symmetrel sollte für Kinder unzugänglich aufbewahrt werden.
Was ist Synthroid 200mcg und wie wird es angewendet?
Synthroid 125 mcg ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome einer Hypothyreose und einer vergrößerten Schilddrüse (Kropf). Synthroid 75mcg kann allein oder mit anderen Medikamenten verwendet werden.
Was sind die möglichen Nebenwirkungen von Synthroid 25mcg?
Synthroid 25 mcg kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Die häufigsten Nebenwirkungen von Synthroid sind:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Synthroid. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
WARNUNG
Schilddrüsenhormone, einschließlich SYNTHROID 125 mcg, sollten weder allein noch mit anderen Therapeutika zur Behandlung von Fettleibigkeit oder zur Gewichtsabnahme verwendet werden. Bei euthyreoten Patienten sind Dosen im Bereich des täglichen Hormonbedarfs zur Gewichtsreduktion unwirksam. Größere Dosen können schwerwiegende oder sogar lebensbedrohliche Toxizitätserscheinungen hervorrufen, insbesondere wenn sie zusammen mit sympathomimetischen Aminen verabreicht werden, wie sie wegen ihrer anorektischen Wirkung verwendet werden.
BEZEICHNUNG
SYNTHROID (Levothyroxin-Natrium-Tabletten, USP) enthält synthetisches kristallines L-3,3',5,5'-Tetraiodthyronin-Natriumsalz [Levothyroxin (T4)-Natrium]. Synthetisches T4 ist identisch mit dem, das in der menschlichen Schilddrüse produziert wird. Levothyroxin (T4)-Natrium hat eine empirische Formel von C15H10I4N NaO4·H2O, das Molekulargewicht von 798,86 g/mol (wasserfrei) und die Strukturformel wie gezeigt:
Inaktive Zutaten
Akazie, Puderzucker (enthält Maisstärke), Lactose-Monohydrat, Magnesiumstearat, Povidon und Talk. Im Folgenden sind die Farbzusätze nach Tablettenstärke aufgeführt:
INDIKATIONEN
Hypothyreose
SYNTHROID ist als Ersatztherapie bei primärer (Schilddrüse), sekundärer (Hypophyse) und tertiärer (Hypothalamus) angeborener oder erworbener Hypothyreose indiziert.
Hypophysen-Thyrotropin (Thyroid-stimulierendes Hormon, TSH) Unterdrückung
SYNTHROID ist als Ergänzung zu einer Operation und Radiojodtherapie bei der Behandlung von thyrotropinabhängigem, gut differenziertem Schilddrüsenkrebs indiziert.
Nutzungsbeschränkungen
DOSIERUNG UND ANWENDUNG
Allgemeine Verwaltungsinformationen
SYNTHROID 25 mcg als tägliche Einzeldosis auf nüchternen Magen eine halbe bis eine Stunde vor dem Frühstück verabreichen.
Verabreichen Sie SYNTHROID mindestens 4 Stunden vor oder nach Arzneimitteln, von denen bekannt ist, dass sie die Resorption von SYNTHROID beeinträchtigen [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Bewerten Sie die Notwendigkeit einer Dosisanpassung, wenn Sie regelmäßig innerhalb einer Stunde bestimmte Nahrungsmittel verabreichen, die die Resorption von SYNTHROID 50 mcg beeinflussen können [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ].
Verabreichen Sie SYNTHROID an Säuglinge und Kinder, die keine ganzen Tabletten schlucken können, indem Sie die Tablette zerdrücken, die frisch zerkleinerte Tablette in einer kleinen Menge (5 bis 10 ml oder 1 bis 2 Teelöffel) Wasser suspendieren und die Suspension sofort mit einem Löffel oder einer Pipette verabreichen. Die Suspension nicht aufbewahren. Nicht in Nahrungsmitteln verabreichen, die die Resorption von SYNTHROID verringern, wie z. B. Säuglingsanfangsnahrung auf Sojabohnenbasis [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Allgemeine Prinzipien der Dosierung
Die Dosis von SYNTHROID 75 mcg bei Hypothyreose oder hypophysärer TSH-Unterdrückung hängt von einer Vielzahl von Faktoren ab, darunter: Alter des Patienten, Körpergewicht, Herz-Kreislauf-Status, Begleiterkrankungen (einschließlich Schwangerschaft), Begleitmedikation, gleichzeitig eingenommene Nahrung und die Spezifische Art der zu behandelnden Erkrankung [vgl Dosierung bei bestimmten Patientenpopulationen , WARNUNGEN UND VORSICHTSMASSNAHMEN , und WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ]. Die Dosierung muss individuell angepasst werden, um diese Faktoren zu berücksichtigen, und Dosisanpassungen müssen auf der Grundlage einer regelmäßigen Beurteilung des klinischen Ansprechens des Patienten und der Laborparameter vorgenommen werden [siehe Überwachung der TSH- und/oder Thyroxin (T4)-Spiegel ].
Die maximale therapeutische Wirkung einer gegebenen SYNTHROID-Dosis wird möglicherweise erst nach 4 bis 6 Wochen erreicht.
Dosierung bei bestimmten Patientenpopulationen
Primäre Hypothyreose bei Erwachsenen und Jugendlichen, bei denen Wachstum und Pubertät abgeschlossen sind
Beginnen Sie mit SYNTHROID mit der vollen Ersatzdosis bei ansonsten gesunden, nicht älteren Personen, die erst seit kurzer Zeit (z. B. einige Monate) an Hypothyreose leiden. Die durchschnittliche vollständige Ersatzdosis von SYNTHROID 125 mcg beträgt ungefähr 1,6 mcg pro kg pro Tag (zum Beispiel: 100 bis 125 mcg pro Tag für einen 70 kg schweren Erwachsenen).
Passen Sie die Dosis alle 4 bis 6 Wochen in Schritten von 12,5 bis 25 mcg an, bis der Patient klinisch euthyreot ist und das Serum-TSH wieder normal ist. Dosen von mehr als 200 mcg pro Tag sind selten erforderlich. Eine unzureichende Reaktion auf Tagesdosen von mehr als 300 mcg pro Tag ist selten und kann auf eine schlechte Compliance, Malabsorption, Arzneimittelwechselwirkungen oder eine Kombination dieser Faktoren hinweisen.
Beginnen Sie bei älteren Patienten oder Patienten mit zugrunde liegender Herzerkrankung mit einer Dosis von 12,5 bis 25 mcg pro Tag. Erhöhen Sie die Dosis je nach Bedarf alle 6 bis 8 Wochen, bis der Patient klinisch euthyreot ist und das Serum-TSH wieder normal ist. Die vollständige Ersatzdosis von SYNTHROID 75 µg kann bei älteren Patienten weniger als 1 µg pro kg pro Tag betragen.
Beginnen Sie bei Patienten mit schwerer, langjähriger Hypothyreose mit einer Dosis von 12,5 bis 25 Mikrogramm pro Tag. Passen Sie die Dosis alle 2 bis 4 Wochen in Schritten von 12,5 bis 25 mcg an, bis der Patient klinisch euthyreot ist und sich der Serum-TSH-Spiegel normalisiert hat.
Sekundäre oder tertiäre Hypothyreose
Beginnen Sie mit SYNTHROID bei ansonsten gesunden, nicht älteren Personen mit der vollen Ersatzdosis. Beginnen Sie mit einer niedrigeren Dosis bei älteren Patienten, Patienten mit zugrunde liegender kardiovaskulärer Erkrankung oder Patienten mit schwerer, langjähriger Hypothyreose, wie oben beschrieben. Serum-TSH ist kein zuverlässiges Maß für die Angemessenheit der Dosis von SYNTHROID 125 µg bei Patienten mit sekundärer oder tertiärer Hypothyreose und sollte nicht zur Überwachung der Therapie verwendet werden. Verwenden Sie den Gehalt an serumfreiem T4, um die Angemessenheit der Therapie bei dieser Patientenpopulation zu überwachen. Titrieren Sie die SYNTHROID-Dosierung gemäß den obigen Anweisungen, bis der Patient klinisch euthyreot ist und der Serumspiegel von freiem T4 auf die obere Hälfte des Normalbereichs zurückgekehrt ist.
Pädiatrische Dosierung – Angeborene oder erworbene Hypothyreose
Die empfohlene Tagesdosis von SYNTHROID bei pädiatrischen Patienten mit Hypothyreose basiert auf dem Körpergewicht und altersbedingten Veränderungen, wie in Tabelle 1 beschrieben. Beginnen Sie mit SYNTHROID bei den meisten pädiatrischen Patienten mit der vollen Tagesdosis. Beginnen Sie mit einer niedrigeren Anfangsdosis bei Neugeborenen (0-3 Monate) mit Risiko für Herzinsuffizienz und bei Kindern mit Risiko für Hyperaktivität (siehe unten). Überwachen Sie das klinische Ansprechen und das Ansprechen im Labor [siehe DOSIERUNG UND ANWENDUNG ].
Neugeborene (0-3 Monate) mit Risiko für Herzinsuffizienz
Erwägen Sie eine niedrigere Anfangsdosis bei Neugeborenen mit einem Risiko für Herzinsuffizienz.
Erhöhen Sie die Dosis je nach klinischem und laborchemischem Ansprechen alle 4 bis 6 Wochen nach Bedarf.
Hyperaktivitätsgefährdete Kinder
Um das Risiko einer Hyperaktivität bei Kindern zu minimieren, beginnen Sie mit einem Viertel der empfohlenen vollen Ersatzdosis und erhöhen Sie diese wöchentlich um ein Viertel der vollen empfohlenen Ersatzdosis, bis die volle empfohlene Ersatzdosis erreicht ist.
Schwangerschaft
Vorbestehende Hypothyreose
Die Dosis von SYNTHROID 200 µg kann während der Schwangerschaft ansteigen. Messen Sie Serum-TSH und freies T4, sobald die Schwangerschaft bestätigt ist, und mindestens während jedes Trimesters der Schwangerschaft. Halten Sie bei Patientinnen mit primärer Hypothyreose das Serum-TSH im Trimester-spezifischen Referenzbereich. Bei Patienten mit Serum-TSH oberhalb des normalen trimenonspezifischen Bereichs die Dosis von SYNTHROID 200 µg um 12,5 bis 25 µg/Tag erhöhen und TSH alle 4 Wochen messen, bis eine stabile SYNTHROID-Dosis erreicht ist und das Serum-TSH im normalen trimenonspezifischen Bereich liegt . Reduzieren Sie die Dosierung von SYNTHROID 25 mcg unmittelbar nach der Entbindung auf die Werte vor der Schwangerschaft und messen Sie die Serum-TSH-Spiegel 4 bis 8 Wochen nach der Geburt, um sicherzustellen, dass die SYNTHROID-Dosis angemessen ist.
Neu auftretende Hypothyreose
Normalisieren Sie die Schilddrüsenfunktion so schnell wie möglich. Beginnen Sie bei Patienten mit mittelschweren bis schweren Anzeichen und Symptomen einer Hypothyreose mit SYNTHROID mit der vollen Ersatzdosis (1,6 µg pro kg Körpergewicht pro Tag). Bei Patienten mit leichter Hypothyreose (TSH Verwendung in bestimmten Bevölkerungsgruppen ].
TSH-Unterdrückung bei gut differenziertem Schilddrüsenkrebs
Im Allgemeinen wird TSH auf unter 0,1 IE pro Liter gesenkt, und dies erfordert normalerweise eine Dosis von SYNTHROID 125 mcg von mehr als 2 mcg pro kg pro Tag. Bei Patienten mit Hochrisikotumoren kann der Zielwert für die TSH-Suppression jedoch niedriger sein.
Überwachung der TSH- und/oder Thyroxin (T4)-Spiegel
Beurteilen Sie die Angemessenheit der Therapie durch regelmäßige Beurteilung von Labortests und klinische Bewertung. Anhaltende klinische und laborchemische Anzeichen einer Hypothyreose trotz einer scheinbar angemessenen Ersatzdosis von SYNTHROID 100 mcg können ein Hinweis auf unzureichende Resorption, schlechte Therapietreue, Arzneimittelwechselwirkungen oder eine Kombination dieser Faktoren sein.
Erwachsene
Bei erwachsenen Patienten mit primärer Hypothyreose sollten die Serum-TSH-Spiegel 6 bis 8 Wochen nach jeder Dosisänderung überwacht werden. Bewerten Sie bei Patienten mit einer stabilen und angemessenen Ersatzdosis das klinische und biochemische Ansprechen alle 6 bis 12 Monate und immer dann, wenn sich der klinische Zustand des Patienten ändert.
Pädiatrie
Beurteilen Sie bei Patienten mit angeborener Hypothyreose die Angemessenheit der Substitutionstherapie, indem Sie sowohl Serum-TSH als auch Gesamt- oder freies T4 messen. Überwachen Sie TSH und Gesamt- oder freies T4 bei Kindern wie folgt: 2 und 4 Wochen nach Beginn der Behandlung, 2 Wochen nach jeder Dosisänderung und danach alle 3 bis 12 Monate nach Dosisstabilisierung, bis das Wachstum abgeschlossen ist. Schlechte Compliance oder anormale Werte können eine häufigere Überwachung erforderlich machen. Führen Sie in regelmäßigen Abständen klinische Routineuntersuchungen durch, einschließlich der Beurteilung der Entwicklung, des geistigen und körperlichen Wachstums und der Knochenreifung.
Während das allgemeine Ziel der Therapie darin besteht, den Serum-TSH-Spiegel zu normalisieren, normalisiert sich TSH bei manchen Patienten aufgrund einer in utero-Hypothyreose möglicherweise nicht, was zu einem Zurücksetzen des Hypophysen-Schilddrüsen-Feedbacks führt. Wenn der Serum-T4-Wert innerhalb von 2 Wochen nach Beginn der SYNTHROID-Therapie nicht in die obere Hälfte des Normalbereichs ansteigt und/oder der Serum-TSH-Wert innerhalb von 4 Wochen unter 20 IE pro Liter abfällt, kann dies darauf hindeuten, dass das Kind keine angemessene Therapie erhält. Beurteilen Sie die Compliance, die Dosis des verabreichten Medikaments und die Verabreichungsmethode, bevor Sie die Dosis von SYNTHROID erhöhen [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und Verwendung in bestimmten Bevölkerungsgruppen ].
Sekundäre und tertiäre Hypothyreose
Die Serumspiegel von freiem T4 überwachen und bei diesen Patienten in der oberen Hälfte des Normalbereichs halten.
WIE GELIEFERT
Darreichungsformen und Stärken
SYNTHROID-Tabletten sind wie folgt erhältlich:
Lagerung und Handhabung
SYNTHROID (Levothyroxin-Natrium, USP) Tabletten werden wie folgt geliefert:
Lagerbedingungen
Bei 25 °C (77 °F) lagern; Exkursionen bis 15° bis 30° C (59° bis 86° F) erlaubt [siehe USP kontrollierte Raumtemperatur ]. SYNTHROID 75 mcg Tabletten sollten vor Licht und Feuchtigkeit geschützt werden.
AbbVie Inc., North Chicago, IL 60064, USA Überarbeitet: Juli 2020
NEBENWIRKUNGEN
Nebenwirkungen im Zusammenhang mit der Therapie mit SYNTHROID 50 µg sind in erster Linie die einer Hyperthyreose aufgrund einer therapeutischen Überdosierung [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN , ÜBERDOSIERUNG ]. Dazu gehören:
Unter der Einleitung einer Levothyroxin-Therapie wurde selten über Krampfanfälle berichtet.
Nebenwirkungen bei Kindern
Bei Kindern, die mit Levothyroxin behandelt wurden, wurde über Pseudotumor cerebri und ein Verrutschen der femoralen Epiphyse berichtet. Eine Überbehandlung kann bei Säuglingen zu einer Kraniosynostose und bei Kindern zu einem vorzeitigen Verschluss der Epiphysen mit daraus resultierender Beeinträchtigung der Erwachsenengröße führen.
Überempfindlichkeitsreaktionen
Bei Patienten, die mit Schilddrüsenhormonpräparaten behandelt wurden, sind Überempfindlichkeitsreaktionen auf inaktive Bestandteile aufgetreten. Dazu gehören Urtikaria, Pruritus, Hautausschlag, Flush, Angioödem, verschiedene gastrointestinale Symptome (Bauchschmerzen, Übelkeit, Erbrechen und Durchfall), Fieber, Arthralgie, Serumkrankheit und Keuchen. Eine Überempfindlichkeit gegenüber Levothyroxin selbst ist nicht bekannt.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Medikamente, von denen bekannt ist, dass sie die Pharmakokinetik von Schilddrüsenhormonen beeinflussen
Viele Arzneimittel können Auswirkungen auf die Pharmakokinetik und den Metabolismus von Schilddrüsenhormonen haben (z. B. Absorption, Synthese, Sekretion, Katabolismus, Proteinbindung und Reaktion des Zielgewebes) und können die therapeutische Reaktion auf SYNTHROID verändern (siehe Tabellen 2-5 unten).
Antidiabetische Therapie
Eine zusätzliche Behandlung mit SYNTHROID 125 µg bei Patienten mit Diabetes mellitus kann die glykämische Kontrolle verschlechtern und zu einem erhöhten Bedarf an Antidiabetika oder Insulin führen. Überwachen Sie die glykämische Kontrolle sorgfältig, insbesondere wenn eine Schilddrüsentherapie begonnen, geändert oder beendet wird [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
Orale Antikoagulanzien
SYNTHROID verstärkt das Ansprechen auf eine orale Antikoagulanzientherapie. Daher kann eine Verringerung der Dosis des Antikoagulans bei Korrektur der Hypothyreose oder bei Erhöhung der SYNTHROID-Dosis von 25 µg gerechtfertigt sein. Gerinnungstests genau überwachen, um angemessene und rechtzeitige Dosisanpassungen zu ermöglichen.
Digitalis-Glykoside
SYNTHROID 125 mcg kann die therapeutische Wirkung von Digitalisglykosiden verringern. Die Digitalisglykoside im Serum können sinken, wenn ein hypothyreoter Patient euthyreot wird, was eine Erhöhung der Digitalisglykoside-Dosis erforderlich macht.
Antidepressive Therapie
Die gleichzeitige Anwendung von trizyklischen (z. B. Amitriptylin) oder tetrazyklischen (z. B. Maprotilin) Antidepressiva und SYNTHROID 25 mcg kann die therapeutischen und toxischen Wirkungen beider Arzneimittel verstärken, möglicherweise aufgrund einer erhöhten Rezeptorempfindlichkeit gegenüber Katecholaminen. Toxische Wirkungen können ein erhöhtes Risiko für Herzrhythmusstörungen und eine Stimulation des zentralen Nervensystems umfassen. SYNTHROID 200 mcg kann den Wirkungseintritt von Trizyklika beschleunigen. Die Verabreichung von Sertralin bei Patienten, die mit SYNTHROID 100 µg stabilisiert sind, kann zu einem erhöhten SYNTHROID-Bedarf führen.
Ketamin
Die gleichzeitige Anwendung von Ketamin und SYNTHROID 50 mcg kann zu deutlichem Bluthochdruck und Tachykardie führen. Bei diesen Patienten sind Blutdruck und Herzfrequenz engmaschig zu überwachen.
Sympathomimetika
Die gleichzeitige Anwendung von Sympathomimetika und SYNTHROID kann die Wirkung von Sympathomimetika oder Schilddrüsenhormon verstärken. Schilddrüsenhormone können das Risiko einer koronaren Insuffizienz erhöhen, wenn Sympathomimetika Patienten mit koronarer Herzkrankheit verabreicht werden.
Tyrosinkinase-Inhibitoren
Die gleichzeitige Anwendung von Tyrosinkinase-Hemmern wie Imatinib kann eine Hypothyreose verursachen. Bei solchen Patienten ist der TSH-Spiegel genau zu überwachen.
Arzneimittel-Lebensmittel-Wechselwirkungen
Der Verzehr bestimmter Nahrungsmittel kann die Resorption von SYNTHROID 25 mcg beeinträchtigen, wodurch eine Anpassung der Dosierung erforderlich wird [siehe DOSIERUNG UND ANWENDUNG ]. Sojabohnenmehl, Baumwollsamenmehl, Walnüsse und Ballaststoffe können SYNTHROID 200 mcg aus dem Magen-Darm-Trakt binden und dessen Aufnahme verringern. Grapefruitsaft kann die Resorption von Levothyroxin verzögern und seine Bioverfügbarkeit verringern.
Arzneimittel-Labortest-Wechselwirkungen
Berücksichtigen Sie bei der Interpretation der T4- und T3-Werte Änderungen der TBG-Konzentration. Messen und bewerten Sie unter diesen Umständen ungebundenes (freies) Hormon und/oder bestimmen Sie den freien T4-Index (FT4I). Schwangerschaft, infektiöse Hepatitis, Östrogene, östrogenhaltige orale Kontrazeptiva und akute intermittierende Porphyrie erhöhen die TBG-Konzentration. Nephrose, schwere Hypoproteinämie, schwere Lebererkrankung, Akromegalie, Androgene und Kortikosteroide verringern die TBG-Konzentration. Familiäre Hyper- oder Hypo-Thyroxin-bindende Globulinämien wurden beschrieben, wobei die Inzidenz eines TBG-Mangels etwa 1 zu 9000 beträgt.
WARNUNGEN
Eingeschlossen als Teil der VORSICHTSMASSNAHMEN Sektion.
VORSICHTSMASSNAHMEN
Kardiale Nebenwirkungen bei älteren Menschen und bei Patienten mit zugrunde liegender Herz-Kreislauf-Erkrankung
Eine Überbehandlung mit Levothyroxin kann eine Zunahme der Herzfrequenz, der Herzwanddicke und der Herzkontraktilität verursachen und Angina pectoris oder Arrhythmien auslösen, insbesondere bei Patienten mit Herz-Kreislauf-Erkrankungen und bei älteren Patienten. Beginnen Sie die SYNTHROID-Therapie bei dieser Population mit niedrigeren Dosen als denen, die für jüngere Personen oder Patienten ohne Herzerkrankung empfohlen werden [siehe DOSIERUNG UND ANWENDUNG , Verwendung in bestimmten Bevölkerungsgruppen ].
Überwachen Sie Herzrhythmusstörungen während chirurgischer Eingriffe bei Patienten mit koronarer Herzkrankheit, die eine suppressive SYNTHROID-Therapie erhalten. Überwachen Sie Patienten, die gleichzeitig SYNTHROID 125 µg und Sympathomimetika erhalten, auf Anzeichen und Symptome einer koronaren Insuffizienz.
Wenn kardiale Symptome auftreten oder sich verschlimmern, reduzieren Sie die SYNTHROID-Dosis oder setzen Sie die Behandlung eine Woche lang aus und beginnen Sie mit einer niedrigeren Dosis.
Myxödem Koma
Myxödem-Koma ist ein lebensbedrohlicher Notfall, der durch schlechte Durchblutung und Hypometabolismus gekennzeichnet ist und zu einer unvorhersehbaren Resorption von Levothyroxin-Natrium aus dem Gastrointestinaltrakt führen kann. Die Anwendung von oralen Schilddrüsenhormon-Arzneimitteln zur Behandlung von Myxödem-Koma wird nicht empfohlen. Verabreichen Sie Schilddrüsenhormonprodukte, die für die intravenöse Verabreichung formuliert sind, um Myxödemkoma zu behandeln.
Akute Nebennierenkrise bei Patienten mit gleichzeitiger Nebenniereninsuffizienz
Schilddrüsenhormon erhöht die metabolische Clearance von Glukokortikoiden. Der Beginn einer Schilddrüsenhormontherapie vor Beginn einer Glukokortikoidtherapie kann bei Patienten mit Nebenniereninsuffizienz eine akute Nebennierenkrise auslösen. Behandeln Sie Patienten mit Nebenniereninsuffizienz mit Ersatz-Glukokortikoiden, bevor Sie die Behandlung mit SYNTHROID beginnen [siehe KONTRAINDIKATIONEN ].
Prävention von Hyperthyreose oder unvollständige Behandlung von Hypothyreose
SYNTHROID 100 mcg hat eine enge therapeutische Breite. Eine Über- oder Unterbehandlung mit SYNTHROID 75 mcg kann negative Auswirkungen auf Wachstum und Entwicklung, Herz-Kreislauf-Funktion, Knochenstoffwechsel, Fortpflanzungsfunktion, kognitive Funktion, emotionalen Zustand, Magen-Darm-Funktion sowie Glukose- und Fettstoffwechsel haben. Titrieren Sie die Dosis von SYNTHROID sorgfältig und überwachen Sie die Reaktion auf die Titration, um diese Wirkungen zu vermeiden [siehe DOSIERUNG UND ANWENDUNG ]. Überwachen Sie bei der Anwendung von SYNTHROID 25 µg das Vorhandensein von Arzneimittel- oder Lebensmittelwechselwirkungen und passen Sie die Dosis nach Bedarf an [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN und KLINISCHE PHARMAKOLOGIE ].
Verschlechterung der diabetischen Kontrolle
Eine zusätzliche Behandlung mit Levothyroxin bei Patienten mit Diabetes mellitus kann die glykämische Kontrolle verschlechtern und zu einem erhöhten Bedarf an Antidiabetika oder Insulin führen. Überwachen Sie die glykämische Kontrolle sorgfältig, nachdem Sie SYNTHROID begonnen, gewechselt oder abgesetzt haben [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Verringerte Knochenmineraldichte im Zusammenhang mit Schilddrüsenhormon-Überersatz
Erhöhte Knochenresorption und verringerte Knochenmineraldichte können als Folge einer übermäßigen Levothyroxin-Ersetzung auftreten, insbesondere bei postmenopausalen Frauen. Die erhöhte Knochenresorption kann mit erhöhten Serumspiegeln und einer Ausscheidung von Calcium und Phosphor im Urin, Erhöhungen der alkalischen Knochenphosphatase und unterdrückten Parathormonspiegeln im Serum einhergehen. Verabreichen Sie die minimale Dosis von SYNTHROID, die die gewünschte klinische und biochemische Reaktion erzielt, um dieses Risiko zu mindern.
Nichtklinische Toxikologie
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Es wurden keine Standard-Tierstudien durchgeführt, um das karzinogene Potenzial, das mutagene Potenzial oder die Auswirkungen auf die Fertilität von Levothyroxin zu bewerten.
Verwendung in bestimmten Bevölkerungsgruppen
Schwangerschaft
Zusammenfassung der Risiken
Erfahrungen mit der Anwendung von Levothyroxin bei schwangeren Frauen, einschließlich Daten aus Post-Marketing-Studien, haben keine erhöhten Raten schwerer Geburtsfehler oder Fehlgeburten gemeldet [siehe Daten ]. Mit einer unbehandelten Hypothyreose in der Schwangerschaft sind Risiken für Mutter und Fötus verbunden. Da die TSH-Spiegel während der Schwangerschaft ansteigen können, sollte TSH überwacht und die Dosierung von SYNTHROID während der Schwangerschaft angepasst werden [siehe Klinische Überlegungen ]. Es wurden keine tierexperimentellen Studien mit Levothyroxin während der Schwangerschaft durchgeführt. SYNTHROID sollte während der Schwangerschaft nicht abgesetzt werden und eine während der Schwangerschaft diagnostizierte Hypothyreose sollte umgehend behandelt werden.
Das geschätzte Hintergrundrisiko schwerer Geburtsfehler und Fehlgeburten für die angegebene Population ist unbekannt. In der US-Allgemeinbevölkerung liegt das geschätzte Hintergrundrisiko für schwere Geburtsfehler und Fehlgeburten bei klinisch erkannten Schwangerschaften bei 2 bis 4 % bzw. 15 bis 20 %.
Klinische Überlegungen
Krankheitsbedingtes mütterliches und/oder embryonale/fetales Risiko
Eine mütterliche Hypothyreose während der Schwangerschaft ist mit einer höheren Komplikationsrate verbunden, einschließlich Spontanabort, Schwangerschaftshypertonie, Präeklampsie, Totgeburt und Frühgeburt. Eine unbehandelte mütterliche Hypothyreose kann die neurokognitive Entwicklung des Fötus beeinträchtigen.
Dosisanpassungen während der Schwangerschaft und der Zeit nach der Geburt
Eine Schwangerschaft kann den Bedarf an SYNTHROID erhöhen. Während der Schwangerschaft sollten die Serum-TSH-Spiegel überwacht und die Dosierung von SYNTHROID angepasst werden. Da die TSH-Werte nach der Geburt den Werten vor der Empfängnis ähneln, sollte die SYNTHROID-Dosierung unmittelbar nach der Entbindung wieder auf die Dosis vor der Schwangerschaft zurückkehren [siehe DOSIERUNG UND ANWENDUNG ].
Daten
Menschliche Daten
Levothyroxin ist als Ersatztherapie für Hypothyreose zugelassen. Es gibt eine lange Erfahrung mit der Anwendung von Levothyroxin bei Schwangeren, einschließlich Daten aus Post-Marketing-Studien, die keine erhöhten Raten fötaler Missbildungen, Fehlgeburten oder anderer unerwünschter mütterlicher oder fötaler Folgen im Zusammenhang mit der Anwendung von Levothyroxin bei schwangeren Frauen berichtet haben.
Stillzeit
Zusammenfassung der Risiken
Begrenzte veröffentlichte Studien berichten, dass Levothyroxin in der Muttermilch vorhanden ist. Es liegen jedoch keine ausreichenden Informationen vor, um die Auswirkungen von Levothyroxin auf den gestillten Säugling zu bestimmen, und es liegen keine Informationen zu den Auswirkungen von Levothyroxin auf die Milchproduktion vor. Eine angemessene Behandlung mit Levothyroxin während der Stillzeit kann die Milchproduktion bei stillenden Müttern mit Schilddrüsenunterfunktion normalisieren. Die Entwicklungs- und Gesundheitsvorteile des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an SYNTHROID und möglichen Nebenwirkungen von SYNTHROID 50 mcg auf den gestillten Säugling oder von der zugrunde liegenden Erkrankung der Mutter berücksichtigt werden.
Pädiatrische Verwendung
Die Anfangsdosis von SYNTHROID variiert mit Alter und Körpergewicht. Dosisanpassungen basieren auf einer Beurteilung der klinischen und Laborparameter des einzelnen Patienten [siehe DOSIERUNG UND ANWENDUNG ].
Bei Kindern, bei denen keine Diagnose einer permanenten Hypothyreose gestellt wurde, sollte die Verabreichung von SYNTHROID 200 mcg für einen Probezeitraum unterbrochen werden, jedoch erst, wenn das Kind mindestens 3 Jahre alt ist. Ermitteln Sie am Ende des Versuchszeitraums die Serum-T4- und TSH-Spiegel und verwenden Sie die Labortestergebnisse und die klinische Bewertung, um die Diagnose und Behandlung zu leiten, falls dies gerechtfertigt ist.
Angeborene Hypothyreose
[Sehen DOSIERUNG UND ANWENDUNG ]
Die schnelle Wiederherstellung normaler T4-Konzentrationen im Serum ist wesentlich, um die nachteiligen Auswirkungen einer angeborenen Hypothyreose auf die intellektuelle Entwicklung sowie auf das allgemeine körperliche Wachstum und die Reifung zu verhindern. Beginnen Sie daher sofort nach der Diagnose mit der Therapie mit SYNTHROID 50 mcg. Levothyroxin wird bei diesen Patienten im Allgemeinen lebenslang verabreicht.
Überwachen Sie Säuglinge während der ersten 2 Wochen der SYNTHROID-Therapie engmaschig auf kardiale Überlastung, Arrhythmien und Aspiration durch eifriges Saugen.
Patienten engmaschig überwachen, um eine Unter- oder Überbehandlung zu vermeiden. Unterbehandlung kann schädliche Auswirkungen auf die intellektuelle Entwicklung und das lineare Wachstum haben. Eine Überbehandlung wird bei Säuglingen mit Kraniosynostose in Verbindung gebracht, kann das Tempo der Gehirnreifung nachteilig beeinflussen und das Knochenalter beschleunigen und zu einem vorzeitigen Epiphysenschluss und einer Beeinträchtigung der Erwachsenengröße führen.
Erworbene Hypothyreose bei pädiatrischen Patienten
Patienten engmaschig überwachen, um Unter- und Überbehandlung zu vermeiden. Eine unzureichende Behandlung kann zu schlechten schulischen Leistungen aufgrund von Konzentrationsstörungen und verlangsamter Denkfähigkeit sowie zu einer verringerten Körpergröße führen. Eine Überbehandlung kann das Knochenalter beschleunigen und zu einem vorzeitigen Epiphysenschluss und einer Beeinträchtigung der Erwachsenengröße führen.
Behandelte Kinder können eine Phase des Aufholwachstums aufweisen, die in einigen Fällen ausreichend sein kann, um die Körpergröße eines Erwachsenen zu normalisieren. Bei Kindern mit schwerer oder anhaltender Hypothyreose reicht das aufholende Wachstum möglicherweise nicht aus, um die Körpergröße eines Erwachsenen zu normalisieren.
Geriatrische Verwendung
Aufgrund der erhöhten Prävalenz von Herz-Kreislauf-Erkrankungen bei älteren Menschen sollte SYNTHROID mit weniger als der vollen Ersatzdosis begonnen werden [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und DOSIERUNG UND ANWENDUNG ]. Vorhofarrhythmien können bei älteren Patienten auftreten. Vorhofflimmern ist die häufigste Arrhythmie, die bei einer Levothyroxin-Überbehandlung bei älteren Patienten beobachtet wird.
ÜBERDOSIS
Die Anzeichen und Symptome einer Überdosierung sind die einer Hyperthyreose [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN und NEBENWIRKUNGEN ]. Außerdem können Verwirrung und Orientierungslosigkeit auftreten. Hirnembolie, Schock, Koma und Tod wurden berichtet. Krampfanfälle traten bei einem 3-jährigen Kind auf, das 3,6 mg Levothyroxin einnahm. Die Symptome sind möglicherweise nicht unbedingt offensichtlich oder treten erst einige Tage nach der Einnahme von Levothyroxin-Natrium auf.
Reduzieren Sie die SYNTHROID-Dosis oder setzen Sie die Behandlung vorübergehend ab, wenn Anzeichen oder Symptome einer Überdosierung auftreten. Entsprechend dem medizinischen Zustand des Patienten eine geeignete unterstützende Behandlung einleiten.
Wenden Sie sich für aktuelle Informationen zum Umgang mit Vergiftungen oder Überdosierungen an das National Poison Control Center unter 1-800-222-1222 oder www.poison.org.
KONTRAINDIKATIONEN
SYNTHROID ist kontraindiziert bei Patienten mit unkorrigierter Nebenniereninsuffizienz [siehe WARNUNGEN UND VORSICHTSMASSNAHMEN ].
KLINISCHE PHARMAKOLOGIE
Wirkmechanismus
Schilddrüsenhormone üben ihre physiologischen Wirkungen durch Kontrolle der DNA-Transkription und Proteinsynthese aus. Trijodthyronin (T3) und L-Thyroxin (T4) diffundieren in den Zellkern und binden an an die DNA gebundene Schilddrüsenrezeptorproteine. Dieser Hormonkernrezeptorkomplex aktiviert die Gentranskription und Synthese von Boten-RNA und zytoplasmatischen Proteinen.
Die physiologischen Wirkungen der Schilddrüsenhormone werden hauptsächlich von T3 produziert, wovon der Großteil (ungefähr 80 %) durch Deiodierung in peripheren Geweben von T4 stammt.
Pharmakodynamik
Orales Levothyroxin-Natrium ist ein synthetisches T4-Hormon, das die gleiche physiologische Wirkung wie endogenes T4 ausübt und dadurch normale T4-Spiegel aufrechterhält, wenn ein Mangel vorliegt.
Pharmakokinetik
Absorption
Die Resorption von oral verabreichtem T4 aus dem Gastrointestinaltrakt reicht von 40 % bis 80 %. Der Großteil der SYNTHROID-Dosis wird aus dem Jejunum und dem oberen Ileum resorbiert. Die relative Bioverfügbarkeit von SYNTHROID 125 µg Tabletten im Vergleich zu einer gleichen Nominaldosis oraler Levothyroxin-Natrium-Lösung beträgt etwa 93 %. Die T4-Resorption wird durch Fasten erhöht und bei Malabsorptionssyndromen und durch bestimmte Lebensmittel wie Sojabohnen verringert. Ballaststoffe verringern die Bioverfügbarkeit von T4. Die Absorption kann auch mit dem Alter abnehmen. Darüber hinaus beeinflussen viele Medikamente und Nahrungsmittel die T4-Absorption [siehe WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ].
Verteilung
Zirkulierende Schilddrüsenhormone sind zu mehr als 99 % an Plasmaproteine gebunden, einschließlich Thyroxin-bindendes Globulin (TBG), Thyroxin-bindendes Präalbumin (TBPA) und Albumin (TBA), deren Kapazitäten und Affinitäten für jedes Hormon variieren. Die höhere Affinität sowohl von TBG als auch von TBPA für T4 erklärt teilweise die höheren Serumspiegel, die langsamere metabolische Clearance und die längere Halbwertszeit von T4 im Vergleich zu T3. Proteingebundene Schilddrüsenhormone stehen im umgekehrten Gleichgewicht mit geringen Mengen an freiem Hormon. Nur ungebundenes Hormon ist metabolisch aktiv. Viele Medikamente und physiologische Zustände beeinflussen die Bindung von Schilddrüsenhormonen an Serumproteine [vgl WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN ]. Schilddrüsenhormone passieren die Plazentaschranke nicht ohne weiteres [siehe Verwendung in bestimmten Bevölkerungsgruppen ].
Beseitigung
Stoffwechsel
T4 wird langsam eliminiert (siehe Tabelle 7). Der Hauptweg des Schilddrüsenhormonstoffwechsels verläuft über die sequentielle Deiodierung. Etwa 80 % des zirkulierenden T3 stammen aus peripherem T4 durch Monodeiodierung. Die Leber ist der Hauptort des Abbaus von T4 und T3, wobei die T4-Dejodierung auch an einer Reihe zusätzlicher Stellen stattfindet, einschließlich der Niere und anderen Geweben. Ungefähr 80 % der täglichen T4-Dosis werden dejodiert, um gleiche Mengen an T3 und reversem T3 (rT3) zu ergeben. T3 und rT3 werden weiter zu Diiodthyronin deiodiert. Schilddrüsenhormone werden auch durch Konjugation mit Glucuroniden und Sulfaten metabolisiert und direkt in die Galle und den Darm ausgeschieden, wo sie einer enterohepatischen Rezirkulation unterliegen.
Ausscheidung
Schilddrüsenhormone werden hauptsächlich über die Nieren ausgeschieden. Ein Teil des konjugierten Hormons gelangt unverändert in den Dickdarm und wird mit dem Kot ausgeschieden. Ungefähr 20 % von T4 werden mit dem Stuhl ausgeschieden. Die Ausscheidung von T4 im Urin nimmt mit dem Alter ab.
INFORMATIONEN ZUM PATIENTEN
Informieren Sie den Patienten über die folgenden Informationen, um die sichere und wirksame Anwendung von SYNTHROID zu unterstützen:
Dosierung und Verabreichung
Wichtige Informationen
Nebenwirkungen
Was ist Atenolol (Tenormin) und wie wird es angewendet?
Atenolol ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Bluthochdruck, Brustschmerzen und bei Patienten nach einem Herzinfarkt. Atenolol kann allein oder mit anderen Medikamenten verwendet werden.
Welche Nebenwirkungen kann Atenolol haben?
Atenolol kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Die häufigsten Nebenwirkungen von Atenolol sind:
Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Atenolol. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
TENORMIN® (Atenolol), ein synthetischer, beta1-selektiver (kardioselektiver) Adrenorezeptorblocker, kann chemisch als Benzolacetamid, 4-[2'-Hydroxy-3'-[(1-Methylethyl)amino beschrieben werden ] propoxy]-. Die Summen- und Strukturformeln lauten:
Atenolol (freie Base) hat ein Molekulargewicht von 266. Es ist eine relativ polare hydrophile Verbindung mit einer Wasserlöslichkeit von 26,5 mg/ml bei 37 °C und einem log-Verteilungskoeffizienten (Octanol/Wasser) von 0,23. Es ist gut löslich in 1 N HCl (300 mg/ml bei 25°C) und weniger löslich in Chloroform (3 mg/ml bei 25°C).
TENORMIN ist als 25-, 50- und 100-mg-Tablette zur oralen Verabreichung erhältlich.
Inaktive Zutaten: Magnesiumstearat, mikrokristalline Cellulose, Povidon, Natriumstärkeglykolat.
Was ist Tenormin 50 mg Injektion und wie wird es angewendet?
Tenormin 25 mg Injektion ist ein verschreibungspflichtiges Arzneimittel zur Behandlung der Symptome von Bluthochdruck, Brustschmerzen und bei Patienten nach einem Herzinfarkt. Tenormin-Injektion kann allein oder mit anderen Medikamenten verwendet werden.
Tenormin 100 mg Injektion ist ein Beta-Blocker, Beta-1-Selektiv.
Was sind die möglichen Nebenwirkungen von Tenormin-Injektion?
Tenormin 25 mg Injektion kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Zu den häufigsten Nebenwirkungen von Tenormin-Injektion gehören:
Informieren Sie Ihren Arzt, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Tenormin Injection. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
TENORMIN® (Atenolol), ein synthetischer, beta1-selektiver (kardioselektiver) Adrenorezeptorblocker, kann chemisch als Benzolacetamid, 4-[2'-Hydroxy-3'-[(1 Methylethyl)amino]propoxy] beschrieben werden. -. Die Summen- und Strukturformeln lauten:
C14H22N2O3
Atenolol (freie Base) hat ein Molekulargewicht von 266. Es ist eine relativ polare hydrophile Verbindung mit einer Wasserlöslichkeit von 26,5 mg/ml bei 37 °C und einem log-Verteilungskoeffizienten (Octanol/Wasser) von 0,23. Es ist gut löslich in 1 N HCl (300 mg/ml bei 25°C) und weniger löslich in Chloroform (3 mg/ml bei 25°C).
TENORMIN zur parenteralen Verabreichung ist als TENORMIN 100 mg IV-Injektion mit 5 mg Atenolol in 10 ml steriler, isotonischer, citratgepufferter, wässriger Lösung erhältlich. Der pH-Wert der Lösung beträgt 5,5–6,5.
Inaktive Zutaten : Natriumchlorid für die Isotonie und Zitronensäure und Natriumhydroxid zur Einstellung des pH-Werts.
INDIKATIONEN
Hypertonie
TENORMIN ist angezeigt zur Behandlung von Bluthochdruck, um den Blutdruck zu senken. Die Senkung des Blutdrucks senkt das Risiko tödlicher und nicht tödlicher kardiovaskulärer Ereignisse, vor allem Schlaganfälle und Myokardinfarkte. Diese Vorteile wurden in kontrollierten Studien mit blutdrucksenkenden Arzneimitteln aus einer Vielzahl von pharmakologischen Klassen, einschließlich Atenolol, beobachtet.
Die Kontrolle des Bluthochdrucks sollte Teil eines umfassenden kardiovaskulären Risikomanagements sein, einschließlich, soweit angemessen, Lipidkontrolle, Diabetesmanagement, antithrombotische Therapie, Raucherentwöhnung, Bewegung und begrenzte Natriumaufnahme. Viele Patienten benötigen mehr als 1 Medikament, um die Blutdruckziele zu erreichen. Spezifische Ratschläge zu Zielen und Management finden Sie in den veröffentlichten Richtlinien, wie denen des Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC) des National High Blood Pressure Education Program.
In randomisierten kontrollierten Studien wurde gezeigt, dass zahlreiche Antihypertensiva aus einer Vielzahl von pharmakologischen Klassen und mit unterschiedlichen Wirkmechanismen die kardiovaskuläre Morbidität und Mortalität verringern, und es kann geschlussfolgert werden, dass es sich um eine Blutdrucksenkung und nicht um eine andere pharmakologische Eigenschaft handelt die Medikamente, die größtenteils für diese Vorteile verantwortlich sind. Der größte und beständigste Vorteil für das kardiovaskuläre Ergebnis war eine Verringerung des Schlaganfallrisikos, aber auch eine Verringerung des Myokardinfarkts und der kardiovaskulären Mortalität wurde regelmäßig beobachtet.
Ein erhöhter systolischer oder diastolischer Druck verursacht ein erhöhtes kardiovaskuläres Risiko, und der absolute Risikoanstieg pro mmHg ist bei höheren Blutdrucken größer, so dass selbst eine geringfügige Verringerung einer schweren Hypertonie einen erheblichen Nutzen bringen kann. Die Reduktion des relativen Risikos durch Blutdrucksenkung ist in Populationen mit unterschiedlichem absolutem Risiko ähnlich, sodass der absolute Nutzen bei Patienten größer ist, die unabhängig von ihrer Hypertonie einem höheren Risiko ausgesetzt sind (z. B. Patienten mit Diabetes oder Hyperlipidämie), und solche Patienten wären zu erwarten um von einer aggressiveren Behandlung zu einem niedrigeren Blutdruckziel zu profitieren.
Einige Antihypertensiva haben geringere Blutdruckwirkungen (als Monotherapie) bei Patienten mit schwarzer Hautfarbe, und viele Antihypertensiva haben zusätzliche zugelassene Indikationen und Wirkungen (z. B. bei Angina pectoris, Herzinsuffizienz oder diabetischer Nierenerkrankung). Diese Überlegungen können die Auswahl der Therapie leiten.
TENORMIN 50 mg kann zusammen mit anderen blutdrucksenkenden Mitteln verabreicht werden.
Angina Pectoris aufgrund von Koronararteriosklerose
TENORMIN ist für die Langzeitbehandlung von Patienten mit Angina pectoris indiziert.
Akuter Myokardinfarkt
TENORMIN ist indiziert zur Behandlung hämodynamisch stabiler Patienten mit gesichertem oder vermutetem akutem Myokardinfarkt, um die kardiovaskuläre Sterblichkeit zu reduzieren. Die Behandlung kann eingeleitet werden, sobald der klinische Zustand des Patienten dies zulässt. (Sehen DOSIERUNG UND ANWENDUNG , KONTRAINDIKATIONEN , und WARNUNGEN .) Im Allgemeinen gibt es keine Grundlage für die Behandlung von Patienten, die aus der ISIS-1-Studie ausgeschlossen wurden (Blutdruck unter 100 mm Hg systolisch, Herzfrequenz unter 50 bpm) oder andere Gründe haben, eine Betablockade zu vermeiden. Wie oben erwähnt, schienen einige Untergruppen (z. B. ältere Patienten mit einem systolischen Blutdruck unter 120 mmHg) weniger wahrscheinlich zu profitieren.
DOSIERUNG UND ANWENDUNG
Hypertonie
Die Anfangsdosis von TENORMIN beträgt 50 mg, verabreicht als eine Tablette täglich, entweder allein oder zusätzlich zu einer diuretischen Therapie. Die volle Wirkung dieser Dosis wird normalerweise innerhalb von ein bis zwei Wochen sichtbar. Wenn kein optimales Ansprechen erzielt wird, sollte die Dosierung auf TENORMIN 100 mg als eine Tablette täglich erhöht werden. Es ist unwahrscheinlich, dass eine Erhöhung der Dosierung über 100 mg pro Tag hinaus einen weiteren Nutzen bringt.
TENORMIN kann allein oder zusammen mit anderen blutdrucksenkenden Mitteln, einschließlich Diuretika vom Thiazidtyp, Hydralazin, Prazosin und Alpha-Methyldopa, angewendet werden.
Angina pectoris
Die Anfangsdosis von TENORMIN 50 mg beträgt 50 mg als eine Tablette täglich. Wenn innerhalb einer Woche kein optimales Ansprechen erzielt wird, sollte die Dosierung auf TENORMIN 100 mg als eine Tablette täglich erhöht werden. Einige Patienten benötigen möglicherweise eine Dosis von 200 mg einmal täglich, um eine optimale Wirkung zu erzielen.
Eine 24-Stunden-Kontrolle bei einmal täglicher Gabe wird erreicht, indem Dosen gegeben werden, die größer als notwendig sind, um eine sofortige maximale Wirkung zu erzielen. Die maximale frühe Wirkung auf die Belastungstoleranz tritt bei Dosen von 50 bis 100 mg auf, aber bei diesen Dosen wird die Wirkung nach 24 Stunden abgeschwächt und beträgt im Durchschnitt etwa 50 % bis 75 % der Wirkung, die bei einmal täglich oralen Dosen von 200 mg beobachtet wird.
Akuter Myokardinfarkt
Bei Patienten mit eindeutigem oder vermutetem akutem Myokardinfarkt sollte die Behandlung mit TENORMIN 25 mg i.v. Injektion so bald wie möglich nach der Ankunft des Patienten im Krankenhaus und nach Feststellung der Eignung begonnen werden. Eine solche Behandlung sollte unmittelbar nach Stabilisierung des hämodynamischen Zustands des Patienten in einer Koronarstation oder einer ähnlichen Abteilung eingeleitet werden. Die Behandlung sollte mit der intravenösen Verabreichung von 5 mg TENORMIN 100 mg über 5 Minuten beginnen, gefolgt von einer weiteren intravenösen Injektion von 5 mg 10 Minuten später. TENORMIN 50 mg i.v. Injektion sollte unter sorgfältig kontrollierten Bedingungen verabreicht werden, einschließlich Überwachung des Blutdrucks, der Herzfrequenz und des Elektrokardiogramms. Es können Verdünnungen von TENORMIN 100 mg IV-Injektion in Dextrose-Injektion USP, Natriumchlorid-Injektion USP oder Natriumchlorid- und Dextrose-Injektion verwendet werden. Diese Zusatzmittel sind 48 Stunden haltbar, wenn sie nicht sofort verwendet werden.
Bei Patienten, die die volle intravenöse Dosis (10 mg) vertragen, sollte die Behandlung mit TENORMIN Tabletten 50 mg 10 Minuten nach der letzten intravenösen Dosis begonnen werden, gefolgt von einer weiteren oralen Dosis von 50 mg 12 Stunden später. Danach kann TENORMIN 50 mg oral entweder 100 mg einmal täglich oder 50 mg zweimal täglich für weitere 6-9 Tage oder bis zur Entlassung aus dem Krankenhaus gegeben werden. Wenn eine behandlungsbedürftige Bradykardie oder Hypotonie oder andere unerwünschte Wirkungen auftreten, sollte TENORMIN abgesetzt werden. (Sehen vollständige Verschreibungsinformationen vor Beginn der Therapie mit TENORMIN-Tabletten .)
Daten aus anderen Betablocker-Studien deuten darauf hin, dass bei Fragen zur intravenösen Anwendung von Betablockern oder einer klinischen Einschätzung, dass eine Kontraindikation vorliegt, der IV-Betablocker eliminiert werden kann und Patienten, die die Sicherheitskriterien erfüllen, TENORMIN Tabletten 50 mg zweimal verabreicht werden können täglich oder 100 mg einmal täglich für mindestens sieben Tage (wenn die IV-Dosierung ausgeschlossen ist).
Obwohl der Wirksamkeitsnachweis von TENORMIN 100 mg vollständig auf Daten aus den ersten sieben Tagen nach dem Infarkt basiert, deuten Daten aus anderen Betablocker-Studien darauf hin, dass die Behandlung mit Betablockern, die nach dem Infarkt wirksam sind, gegebenenfalls ein bis drei Jahre fortgesetzt werden kann keine Kontraindikationen.
TENORMIN ist eine zusätzliche Behandlung zur Standardtherapie auf der Koronarstation.
Ältere Patienten oder Patienten mit eingeschränkter Nierenfunktion
TENORMIN wird über die Nieren ausgeschieden; daher sollte die Dosierung bei schwerer Beeinträchtigung der Nierenfunktion angepasst werden. Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein und normalerweise am unteren Ende des Dosierungsbereichs beginnen, was die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und einer Begleiterkrankung oder einer anderen medikamentösen Therapie widerspiegelt. Die Beurteilung von Patienten mit Bluthochdruck oder Myokardinfarkt sollte immer eine Beurteilung der Nierenfunktion umfassen. Es ist zu erwarten, dass die Atenolol-Ausscheidung mit zunehmendem Alter abnimmt.
Es tritt keine signifikante Akkumulation von TENORMIN 100 mg auf, bis die Kreatinin-Clearance unter 35 ml/min/1,73 m² fällt. Die Akkumulation von Atenolol und die Verlängerung seiner Halbwertszeit wurden bei Patienten mit einer Kreatinin-Clearance zwischen 5 und 105 ml/min untersucht. Die maximalen Plasmaspiegel waren bei Patienten mit einer Kreatinin-Clearance unter 30 ml/min signifikant erhöht.
Die folgenden maximalen oralen Dosierungen werden für ältere Patienten mit eingeschränkter Nierenfunktion und für Patienten mit eingeschränkter Nierenfunktion aufgrund anderer Ursachen empfohlen:
Einige nierengeschädigte oder ältere Patienten, die wegen Bluthochdruck behandelt werden, benötigen möglicherweise eine niedrigere Anfangsdosis von TENORMIN: 25 mg als eine Tablette täglich. Wenn diese 25-mg-Dosis verwendet wird, muss die Wirksamkeit sorgfältig beurteilt werden. Dies sollte die Messung des Blutdrucks unmittelbar vor der nächsten Dosis („Talblutdruck“) umfassen, um sicherzustellen, dass die Behandlungswirkung über volle 24 Stunden anhält.
Obwohl bei älteren und/oder nierengeschädigten Patienten, die wegen anderer Indikationen als Bluthochdruck behandelt werden, eine ähnliche Dosisreduktion in Erwägung gezogen werden kann, liegen für diese Patientengruppen keine Daten vor.
Hämodialysepatienten sollten nach jeder Dialyse 25 mg oder 50 mg erhalten; dies sollte unter Krankenhausaufsicht erfolgen, da ein deutlicher Blutdruckabfall auftreten kann.
Beendigung der Therapie bei Patienten mit Angina pectoris
Wenn ein Absetzen der TENORMIN-Therapie geplant ist, sollte dies schrittweise erreicht werden, und die Patienten sollten sorgfältig überwacht und angewiesen werden, die körperliche Aktivität auf ein Minimum zu beschränken.
WIE GELIEFERT
TENORMIN 50 mg Tabletten
Tabletten mit 25 mg Atenolol, NDC 0310-0107 (runde, flache, unbeschichtete weiße Tabletten, die auf der einen Seite mit „T“ und auf der anderen Seite mit der Prägung „107“ gekennzeichnet sind) werden in Flaschen mit 100 Tabletten geliefert.
Tabletten mit 50 mg Atenolol, NDC 0310-0105 (runde, flache, unbeschichtete weiße Tabletten mit der Prägung „TENORMIN“ auf einer Seite und der Prägung 105 auf der anderen Seite, halbiert) sind in Flaschen mit 100 Tabletten erhältlich.
Tabletten mit 100 mg Atenolol, NDC 0310-0101 (runde, flache, unbeschichtete weiße Tabletten mit der Prägung „TENORMIN“ auf einer Seite und der Prägung 101 auf der anderen Seite) sind in Flaschen mit 100 Tabletten erhältlich.
Bei kontrollierter Raumtemperatur lagern, 20-25°C (68-77°F) [siehe USP ]. In gut verschlossene, lichtbeständige Behälter füllen.
Vertrieb durch: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. Überarbeitet: Okt. 2012
NEBENWIRKUNGEN
Die meisten Nebenwirkungen waren mild und vorübergehend.
Die Häufigkeitsschätzungen in der folgenden Tabelle wurden aus kontrollierten Studien bei Bluthochdruckpatienten abgeleitet, in denen Nebenwirkungen entweder freiwillig vom Patienten (US-Studien) oder z. B. durch eine Checkliste (ausländische Studien) erhoben wurden. Die berichtete Häufigkeit ausgelöster Nebenwirkungen war sowohl bei mit TENORMIN als auch bei mit Placebo behandelten Patienten höher als bei freiwillig gemeldeten Nebenwirkungen. Wo die Häufigkeit von Nebenwirkungen von TENORMIN und Placebo ähnlich ist, ist der kausale Zusammenhang mit TENORMIN 25 mg ungewiss.
Akuter Myokardinfarkt
In einer Reihe von Untersuchungen zur Behandlung des akuten Myokardinfarkts traten Bradykardie und Hypotonie, wie für jeden Betablocker zu erwarten, bei mit Atenolol behandelten Patienten häufiger auf als bei Kontrollpatienten. Diese sprachen jedoch in der Regel auf Atropin und/oder auf das Aussetzen einer weiteren Dosis von Atenolol an. Die Inzidenz von Herzinsuffizienz wurde durch Atenolol nicht erhöht. Inotrope Mittel wurden selten verwendet. Die gemeldete Häufigkeit dieser und anderer Ereignisse, die während dieser Untersuchungen auftraten, ist in der folgenden Tabelle angegeben. In einer Studie mit 477 Patienten wurden die folgenden Nebenwirkungen entweder während der intravenösen und/oder oralen Verabreichung von Atenolol berichtet:
In der anschließenden International Study of Infarct Survival (ISIS-1), an der über 16.000 Patienten teilnahmen, von denen 8.037 randomisiert einer Behandlung mit TENORMIN 50 mg zugeteilt wurden, wurde die Dosierung von intravenösem und anschließendem oralem TENORMIN aus den folgenden Gründen entweder abgesetzt oder reduziert:
Während der Postmarketing-Erfahrung mit TENORMIN 25 mg wurde im zeitlichen Zusammenhang mit der Anwendung des Arzneimittels Folgendes berichtet: Erhöhte Leberenzyme und/oder Bilirubin, Halluzinationen, Kopfschmerzen, Impotenz, Peyronie-Krankheit, orthostatische Hypotonie, die mit Synkopen einhergehen kann, psoriasiformer Hautausschlag oder Exazerbation von Psoriasis, Psychosen, Purpura, reversibler Alopezie, Thrombozytopenie, Sehstörungen, Sick-Sinus-Syndrom und Mundtrockenheit. TENORMIN 50 mg wurde wie andere Betablocker mit der Entwicklung von antinukleären Antikörpern (ANA), dem Lupus-Syndrom und dem Raynaud-Phänomen in Verbindung gebracht.
Mögliche Nebenwirkungen
Darüber hinaus wurde bei anderen Betablockern eine Reihe von Nebenwirkungen berichtet, die als potenzielle Nebenwirkungen von TENORMIN angesehen werden können.
Hämatologisch: Agranulozytose.
Allergisch: Fieber, kombiniert mit Schmerzen und Halsschmerzen, Laryngospasmus und Atemnot.
Zentrales Nervensystem: Reversible mentale Depression, die zu Katatonie fortschreitet; ein akutes reversibles Syndrom, das durch zeitliche und räumliche Desorientierung gekennzeichnet ist; Verlust des Kurzzeitgedächtnisses; emotionale Labilität mit leicht getrübtem Sensorium; und verminderte Leistung bei Neuropsychometrie.
Magen-Darm: Mesenterialarterienthrombose, ischämische Kolitis.
Sonstiges: Erythematöser Ausschlag.
Sonstig: Es gibt Berichte über Hautausschläge und/oder trockene Augen im Zusammenhang mit der Anwendung von Betarezeptorenblockern. Die berichtete Inzidenz ist gering und in den meisten Fällen sind die Symptome nach Absetzen der Behandlung abgeklungen. Ein Absetzen des Arzneimittels sollte in Betracht gezogen werden, wenn eine solche Reaktion nicht anderweitig erklärbar ist. Die Patienten sollten nach Beendigung der Therapie engmaschig überwacht werden. (Sehen DOSIERUNG UND ANWENDUNG .)
Das okulomukokutane Syndrom im Zusammenhang mit dem Betablocker Practolol wurde bei TENORMIN nicht berichtet. Darüber hinaus wurde eine Reihe von Patienten, die zuvor etablierte Practolol-Reaktionen gezeigt hatten, auf eine TENORMIN-Therapie mit anschließendem Abklingen oder Abklingen der Reaktion umgestellt.
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Katecholamin-abbauende Arzneimittel (z. B. Reserpin) können eine additive Wirkung haben, wenn sie zusammen mit Betablockern verabreicht werden. Patienten, die mit TENORMIN 100 mg plus einem Katecholamin-Mangel behandelt werden, sollten daher engmaschig auf Anzeichen von Hypotonie und/oder ausgeprägter Bradykardie, die Schwindel, Synkopen oder orthostatische Hypotonie hervorrufen können, überwacht werden.
Calciumkanalblocker können auch eine additive Wirkung haben, wenn sie zusammen mit TENORMIN gegeben werden (siehe WARNUNGEN ).
Disopyramid ist ein Typ-I-Antiarrhythmikum mit starken negativ inotropen und chronotropen Wirkungen. Disopyramid wurde bei Verabreichung mit Betablockern mit schwerer Bradykardie, Asystolie und Herzinsuffizienz in Verbindung gebracht.
Amiodaron ist ein Antiarrhythmikum mit negativen chronotropen Eigenschaften, die zu denen von Betablockern hinzukommen können.
Betablocker können die Rebound-Hypertonie verschlimmern, die nach dem Absetzen von Clonidin auftreten kann. Bei gleichzeitiger Anwendung der beiden Arzneimittel sollte der Betablocker einige Tage vor dem schrittweisen Absetzen von Clonidin abgesetzt werden. Wenn Clonidin durch eine Betablocker-Therapie ersetzt wird, sollte die Einführung von Betablockern um einige Tage verschoben werden, nachdem die Clonidin-Verabreichung beendet wurde.
Die gleichzeitige Anwendung von Prostaglandin-Synthase-Hemmern, z. B. Indomethacin, kann die blutdrucksenkende Wirkung von Betablockern verringern.
Informationen zur gleichzeitigen Anwendung von Atenolol und Aspirin sind begrenzt. Daten aus mehreren Studien, z. B. TIMI-II, ISIS-2, deuten derzeit nicht auf eine klinische Wechselwirkung zwischen Aspirin und Betablockern bei akutem Myokardinfarkt hin.
Während der Einnahme von Betablockern können Patienten mit einer Vorgeschichte von anaphylaktischen Reaktionen auf eine Vielzahl von Allergenen eine schwerere Reaktion auf wiederholte Provokation haben, entweder versehentlich, diagnostisch oder therapeutisch. Solche Patienten sprechen möglicherweise nicht auf die üblichen Dosen von Epinephrin an, die zur Behandlung der allergischen Reaktion verwendet werden.
Sowohl Digitalisglykoside als auch Betablocker verlangsamen die atrioventrikuläre Überleitung und verringern die Herzfrequenz. Die gleichzeitige Anwendung kann das Risiko einer Bradykardie erhöhen.
WARNUNGEN
Herzversagen
Eine sympathische Stimulation ist notwendig, um die Kreislauffunktion bei dekompensierter Herzinsuffizienz zu unterstützen, und eine Beta-Blockade birgt die potenzielle Gefahr, die myokardiale Kontraktilität weiter zu unterdrücken und eine schwerere Herzinsuffizienz herbeizuführen.
Bei Patienten mit akutem Myokardinfarkt ist eine Herzinsuffizienz, die nicht umgehend und wirksam durch 80 mg intravenöses Furosemid oder eine gleichwertige Therapie kontrolliert werden kann, eine Kontraindikation für die Behandlung mit Betablockern.
Bei Patienten ohne Herzinsuffizienz in der Vorgeschichte
Eine fortgesetzte Depression des Myokards mit Betablockern über einen längeren Zeitraum kann in einigen Fällen zu Herzversagen führen. Beim ersten Anzeichen oder Symptom einer drohenden Herzinsuffizienz sollten die Patienten entsprechend den derzeit empfohlenen Richtlinien behandelt und das Ansprechen genau beobachtet werden. Wenn die Herzinsuffizienz trotz angemessener Behandlung anhält, sollte TENORMIN abgesetzt werden. (Sehen DOSIERUNG UND ANWENDUNG .)
Beendigung der Therapie mit TENORMIN
Patienten mit koronarer Herzkrankheit, die mit TENORMIN behandelt werden, sollte von einem abrupten Absetzen der Therapie abgeraten werden. Schwere Exazerbation der Angina und das Auftreten von Myokardinfarkt und ventrikulären Arrhythmien wurden bei Angina-Patienten nach abruptem Absetzen der Therapie mit Betablockern berichtet. Die letzten beiden Komplikationen können mit oder ohne vorangegangener Exazerbation der Angina pectoris auftreten. Wie bei anderen Betablockern sollten die Patienten, wenn ein Absetzen von TENORMIN 50 mg geplant ist, sorgfältig überwacht und angewiesen werden, die körperliche Aktivität auf ein Minimum zu beschränken. Wenn sich die Angina pectoris verschlimmert oder sich eine akute Koronarinsuffizienz entwickelt, wird empfohlen, die Behandlung mit TENORMIN 50 mg unverzüglich, zumindest vorübergehend, wieder aufzunehmen. Da die koronare Herzkrankheit häufig ist und unerkannt bleiben kann, ist es ratsam, die TENORMIN-Therapie nicht abrupt abzubrechen, selbst bei Patienten, die nur wegen Bluthochdruck behandelt werden. (Sehen DOSIERUNG UND ANWENDUNG .)
Gleichzeitige Anwendung von Kalziumkanalblockern
Bradykardie und Herzblock können auftreten und der linksventrikuläre enddiastolische Druck kann ansteigen, wenn Betablocker zusammen mit Verapamil oder Diltiazem verabreicht werden. Patienten mit vorbestehenden Überleitungsstörungen oder linksventrikulärer Dysfunktion sind besonders anfällig. (Sehen VORSICHTSMASSNAHMEN .)
Bronchospastische Erkrankungen
PATIENTEN MIT BRONCHOSPASTISCHER ERKRANKUNG SOLLTEN IM ALLGEMEINEN KEINE BETA-BLOCKER ERHALTEN. Aufgrund seiner relativen Beta1-Selektivität kann TENORMIN 50 mg jedoch bei Patienten mit bronchospastischen Erkrankungen, die auf andere blutdrucksenkende Behandlungen nicht ansprechen oder diese nicht vertragen, mit Vorsicht angewendet werden. Da die Beta1-Selektivität nicht absolut ist, sollte bei Therapiebeginn mit 50 mg die niedrigstmögliche Dosis von TENORMIN angewendet und ein Beta2-stimulierendes Mittel (Bronchodilatator) zur Verfügung gestellt werden. Wenn die Dosis erhöht werden muss, sollte eine Aufteilung der Dosis erwogen werden, um niedrigere Spitzenwerte im Blut zu erreichen.
Große Operation
Eine chronisch verabreichte Betablocker-Therapie sollte vor einer größeren Operation nicht routinemäßig abgesetzt werden, jedoch kann die beeinträchtigte Fähigkeit des Herzens, auf adrenerge Reflexstimuli zu reagieren, die Risiken einer Vollnarkose und chirurgischer Eingriffe erhöhen.
Diabetes und Hypoglykämie
TENORMIN 25 mg sollte bei Diabetikern mit Vorsicht angewendet werden, wenn ein Betablocker erforderlich ist. Betablocker können eine bei Hypoglykämie auftretende Tachykardie maskieren, aber andere Manifestationen wie Schwindel und Schwitzen werden möglicherweise nicht signifikant beeinflusst. In der empfohlenen Dosierung potenziert TENORMIN 25 mg die insulininduzierte Hypoglykämie nicht und verzögert im Gegensatz zu nichtselektiven Betablockern nicht die Erholung des Blutzuckerspiegels auf normale Werte.
Thyreotoxikose
Eine beta-adrenerge Blockade kann bestimmte klinische Anzeichen (z. B. Tachykardie) einer Hyperthyreose maskieren. Ein abruptes Absetzen der Betablockade könnte einen Schilddrüsensturm auslösen; Daher sollten Patienten mit Verdacht auf Entwicklung einer Thyreotoxikose, bei denen die TENORMIN-Therapie abgesetzt werden soll, engmaschig überwacht werden. (Sehen DOSIERUNG UND ANWENDUNG .)
Unbehandeltes Phäochromozytom
TENORMIN sollte Patienten mit unbehandeltem Phäochromozytom nicht verabreicht werden.
Schwangerschaft und fetale Verletzung
Atenolol kann den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird. Atenolol passiert die Plazentaschranke und erscheint im Nabelschnurblut. Die Verabreichung von Atenolol ab dem zweiten Trimenon der Schwangerschaft wurde mit der Geburt von Säuglingen in Verbindung gebracht, die für das Gestationsalter klein sind. Es wurden keine Studien zur Anwendung von Atenolol im ersten Trimenon durchgeführt und die Möglichkeit einer fetalen Schädigung kann nicht ausgeschlossen werden. Wenn dieses Arzneimittel während der Schwangerschaft angewendet wird oder wenn die Patientin während der Einnahme dieses Arzneimittels schwanger wird, sollte die Patientin über die potenzielle Gefahr für den Fötus informiert werden.
Neugeborene von Müttern, die TENORMIN 100 mg bei der Geburt oder beim Stillen erhalten, können einem Risiko für Hypoglykämie und Bradykardie ausgesetzt sein. Vorsicht ist geboten, wenn TENORMIN 50 mg während der Schwangerschaft oder einer stillenden Frau verabreicht wird. (Sehen VORSICHTSMASSNAHMEN , Stillende Mutter .)
Es wurde gezeigt, dass Atenolol bei Ratten bei Dosen von mindestens 50 mg/kg/Tag oder dem 25-fachen oder mehr der maximal empfohlenen blutdrucksenkenden Dosis beim Menschen einen dosisabhängigen Anstieg der embryo-/fötalen Resorptionen hervorruft.* Obwohl ähnliche Wirkungen nicht beobachtet wurden bei Kaninchen wurde die Verbindung bei Kaninchen in Dosen über 25 mg/kg/Tag oder dem 12,5-Fachen der maximal empfohlenen blutdrucksenkenden Dosis beim Menschen nicht untersucht.*
*Basierend auf der maximalen Dosis von 100 mg/Tag bei einem 50 kg schweren Patienten.
VORSICHTSMASSNAHMEN
Allgemein
Patienten, die bereits einen Betablocker einnehmen, müssen vor der Verabreichung von TENORMIN sorgfältig untersucht werden. Anfangs- und Folgedosierungen von TENORMIN können je nach klinischen Beobachtungen, einschließlich Puls und Blutdruck, nach unten angepasst werden. TENORMIN kann periphere arterielle Durchblutungsstörungen verschlimmern.
Beeinträchtigte Nierenfunktion
Das Medikament sollte bei Patienten mit eingeschränkter Nierenfunktion mit Vorsicht angewendet werden. (Sehen DOSIERUNG UND ANWENDUNG .)
Karzinogenese, Mutagenese, Beeinträchtigung der Fruchtbarkeit
Zwei Langzeitstudien (maximale Dosierungsdauer von 18 oder 24 Monaten) an Ratten und eine Langzeitstudie (maximale Dosierungsdauer von 18 Monaten) an Mäusen, jeweils mit Dosierungen von bis zu 300 mg/kg/Tag oder dem 150-Fachen des Maximums empfohlene blutdrucksenkende Dosis beim Menschen,* deutete nicht auf ein karzinogenes Potenzial von Atenolol hin. Eine dritte (24-monatige) Rattenstudie mit Dosen von 500 und 1.500 mg/kg/Tag (das 250- und 750-fache der empfohlenen Höchstdosis für blutdrucksenkende Mittel beim Menschen*) führte zu einer erhöhten Inzidenz gutartiger Nebennierenmarkstumoren bei Männern und Frauen, bei Mammafibroadenomen Frauen und Adenome des Hypophysenvorderlappens und parafollikuläre Zellkarzinome der Schilddrüse bei Männern. Im Dominant-Letal-Test (Maus), im In-vivo-Zytogenetik-Test (chinesischer Hamster) oder im Ames-Test (S typhimurium) ergaben sich keine Hinweise auf ein mutagenes Potenzial von Atenolol.
Die Fertilität männlicher oder weiblicher Ratten (bewertet bei Dosierungen von bis zu 200 mg/kg/Tag oder dem 100-Fachen der maximal empfohlenen Humandosis*) wurde durch die Verabreichung von Atenolol nicht beeinflusst.
Tiertoxikologie
Chronische Studien mit oralem Atenolol an Tieren haben das Auftreten einer Vakuolisierung von Epithelzellen der Brunner-Drüsen im Zwölffingerdarm sowohl bei männlichen als auch bei weiblichen Hunden bei allen getesteten Atenolol-Dosismengen (beginnend mit 15 mg/kg/Tag oder dem 7,5-Fachen des Maximums) gezeigt empfohlene blutdrucksenkende Dosis beim Menschen*) und eine erhöhte Inzidenz atrialer Degeneration des Herzens männlicher Ratten bei 300, aber nicht 150 mg Atenolol/kg/Tag (das 150- bzw. 75-fache der maximal empfohlenen blutdrucksenkenden Dosis beim Menschen*).
*Basierend auf der maximalen Dosis von 100 mg/Tag bei einem 50 kg schweren Patienten.
Verwendung in der Schwangerschaft
Schwangerschaftskategorie D
Sehen WARNUNGEN - Schwangerschaft und fetale Verletzung .
Stillende Mutter
Atenolol wird im Vergleich zur Konzentration im Plasma in einem Verhältnis von 1,5 zu 6,8 in die Muttermilch ausgeschieden. Vorsicht ist geboten, wenn TENORMIN 50 mg einer stillenden Frau verabreicht wird. Bei gestillten Säuglingen wurde über klinisch signifikante Bradykardie berichtet. Frühgeborene oder Säuglinge mit eingeschränkter Nierenfunktion können eher Nebenwirkungen entwickeln.
Neugeborene von Müttern, die TENORMIN bei der Geburt oder beim Stillen erhalten, können einem Risiko für Hypoglykämie und Bradykardie ausgesetzt sein. Vorsicht ist geboten, wenn TENORMIN 100 mg während der Schwangerschaft oder an eine stillende Frau verabreicht wird (siehe WARNUNGEN , Schwangerschaft und fetale Verletzung ).
Pädiatrische Verwendung
Sicherheit und Wirksamkeit bei pädiatrischen Patienten wurden nicht nachgewiesen.
Geriatrische Verwendung
Bluthochdruck und Angina pectoris aufgrund von Koronararteriosklerose
Klinische Studien mit TENORMIN 25 mg schlossen keine ausreichende Anzahl von Patienten ab 65 Jahren ein, um festzustellen, ob sie anders reagieren als jüngere Probanden. Andere berichtete klinische Erfahrungen haben keine Unterschiede im Ansprechen zwischen älteren und jüngeren Patienten festgestellt. Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein und normalerweise am unteren Ende des Dosierungsbereichs beginnen, um die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und einer Begleiterkrankung oder einer anderen medikamentösen Therapie widerzuspiegeln.
Akuter Myokardinfarkt
Von den 8.037 Patienten mit Verdacht auf akuten Myokardinfarkt, die in der ISIS-1-Studie zu TENORMIN randomisiert wurden (vgl KLINISCHE PHARMAKOLOGIE ), 33 % (2.644) waren 65 Jahre und älter. Es war nicht möglich, signifikante Unterschiede in der Wirksamkeit und Sicherheit zwischen älteren und jüngeren Patienten festzustellen; ältere Patienten mit einem systolischen Blutdruck INDIKATIONEN UND VERWENDUNG ).
Im Allgemeinen sollte die Dosisauswahl für einen älteren Patienten vorsichtig sein und normalerweise am unteren Ende des Dosierungsbereichs beginnen, was die größere Häufigkeit einer verminderten Leber-, Nieren- oder Herzfunktion und einer Begleiterkrankung oder einer anderen medikamentösen Therapie widerspiegelt. Die Beurteilung von Patienten mit Bluthochdruck oder Myokardinfarkt sollte immer eine Beurteilung der Nierenfunktion umfassen.
ÜBERDOSIS
Über eine Überdosierung mit TENORMIN 100 mg wurde bei Patienten berichtet, die eine akute Dosis von bis zu 5 g überlebten. Ein Todesfall wurde bei einem Mann gemeldet, der akut bis zu 10 g eingenommen haben könnte.
Die vorherrschenden Symptome, die nach einer Überdosierung von TENORMIN 50 mg berichtet wurden, sind Lethargie, Störungen des Atemantriebs, Keuchen, Sinuspause und Bradykardie. Darüber hinaus sind kongestive Herzinsuffizienz, Hypotonie, Bronchospasmus und/oder Hypoglykämie häufige Nebenwirkungen, die mit einer Überdosierung von Betablockern einhergehen und die auch bei einer Überdosierung von TENORMIN 50 mg zu erwarten sind.
Die Behandlung einer Überdosierung sollte auf die Entfernung aller nicht resorbierten Arzneimittel durch induziertes Erbrechen, Magenspülung oder Verabreichung von Aktivkohle gerichtet sein. TENORMIN 50 mg kann durch Hämodialyse aus dem allgemeinen Kreislauf entfernt werden. Andere Behandlungsmodalitäten sollten nach Ermessen des Arztes angewendet werden und können umfassen:
BRADYKARDIE: Atropin intravenös. Wenn die Vagusblockade nicht anspricht, geben Sie Isoproterenol vorsichtig. In refraktären Fällen kann ein transvenöser Herzschrittmacher indiziert sein.
HERZBLOCK (ZWEITER ODER DRITTER GRAD): Isoproterenol oder transvenöser Herzschrittmacher.
HERZVERSAGEN: Digitalisieren Sie den Patienten und verabreichen Sie ein Diuretikum. Es wurde berichtet, dass Glukagon nützlich ist.
HYPOTONIE: Vasopressoren wie Dopamin oder Noradrenalin (Levarterenol). Überwachen Sie den Blutdruck kontinuierlich.
Bronchospasmus: Ein Beta2-Stimulans wie Isoproterenol oder Terbutalin und/oder Aminophyllin.
HYPOGLYKÄMIE: Intravenöse Glukose.
Abhängig von der Schwere der Symptome kann die Behandlung eine intensive Unterstützung und Einrichtungen zur Anwendung von Herz- und Atemunterstützung erfordern.
KONTRAINDIKATIONEN
TENORMIN ist kontraindiziert bei Sinusbradykardie, Herzblock größer als 1. Grades, kardiogenem Schock und manifester Herzinsuffizienz. (Sehen WARNUNGEN .)
TENORMIN ist kontraindiziert bei Patienten mit bekannter Überempfindlichkeit gegen Atenolol oder einen der Bestandteile des Arzneimittels.
KLINISCHE PHARMAKOLOGIE
TENORMIN 100 mg ist ein beta1-selektiver (kardioselektiver) beta-adrenerger Rezeptorblocker ohne membranstabilisierende oder intrinsische sympathomimetische (teilweise agonistische) Aktivität. Diese bevorzugte Wirkung ist jedoch nicht absolut, und bei höheren Dosen hemmt TENORMIN Beta2-Adrenorezeptoren, die sich hauptsächlich in der Bronchial- und Gefäßmuskulatur befinden.
Pharmakokinetik und Stoffwechsel
Beim Menschen erfolgt die Resorption einer oralen Dosis schnell und konsistent, aber unvollständig. Ungefähr 50 % einer oralen Dosis werden aus dem Gastrointestinaltrakt resorbiert, der Rest wird unverändert mit den Faeces ausgeschieden. Maximale Blutspiegel werden zwischen zwei (2) und vier (4) Stunden nach der Einnahme erreicht. Im Gegensatz zu Propranolol oder Metoprolol, aber wie Nadolol, unterliegt TENORMIN nur einem geringen oder keinem Metabolismus durch die Leber, und der resorbierte Anteil wird hauptsächlich durch renale Ausscheidung ausgeschieden. Über 85 % einer intravenösen Dosis werden innerhalb von 24 Stunden im Urin ausgeschieden, verglichen mit etwa 50 % bei einer oralen Dosis. TENORMIN 50mg unterscheidet sich von Propranolol auch dadurch, dass nur eine geringe Menge (6%-16%) an Proteine im Plasma gebunden wird. Dieses kinetische Profil führt zu relativ konsistenten Arzneimittelspiegeln im Plasma mit einer ungefähr vierfachen Variation von Patient zu Patient.
Die Eliminationshalbwertszeit von oralem TENORMIN 100 mg beträgt etwa 6 bis 7 Stunden, und es gibt keine Veränderung des kinetischen Profils des Arzneimittels durch chronische Verabreichung. Nach intravenöser Verabreichung werden maximale Plasmaspiegel innerhalb von 5 Minuten erreicht. Rückgänge von Spitzenwerten sind während der ersten 7 Stunden schnell (5- bis 10-fach); danach fallen die Plasmaspiegel mit einer Halbwertszeit ab, die der eines oral verabreichten Arzneimittels ähnlich ist. Nach oraler Gabe von 50 mg oder 100 mg halten sowohl die Betablocker- als auch die blutdrucksenkende Wirkung für mindestens 24 Stunden an. Bei eingeschränkter Nierenfunktion steht die Elimination von TENORMIN 100 mg in engem Zusammenhang mit der glomerulären Filtrationsrate; Eine signifikante Akkumulation tritt auf, wenn die Kreatinin-Clearance unter 35 ml/min/1,73 m² fällt. (Sehen DOSIERUNG UND ANWENDUNG .)
Pharmakodynamik
In standardmäßigen pharmakologischen Tier- oder Humantests wurde die Beta-Adrenorezeptor-blockierende Aktivität von TENORMIN 25 mg nachgewiesen durch: (1) Verringerung der Ruhe- und Belastungsherzfrequenz und des Herzzeitvolumens, (2) Verringerung des systolischen und diastolischen Blutdrucks in Ruhe und unter Belastung , (3) Hemmung von Isoproterenol-induzierter Tachykardie und (4) Verringerung von orthostatischer Reflextachykardie.
Eine signifikante betablockierende Wirkung von TENORMIN, gemessen an der Verringerung der Belastungstachykardie, ist innerhalb einer Stunde nach oraler Verabreichung einer Einzeldosis offensichtlich. Diese Wirkung ist nach etwa 2 bis 4 Stunden maximal und hält mindestens 24 Stunden an. Die maximale Verringerung der Belastungstachykardie tritt innerhalb von 5 Minuten nach einer intravenösen Dosis auf. Sowohl bei oral als auch intravenös verabreichtem Arzneimittel ist die Wirkungsdauer dosisabhängig und steht auch in linearer Beziehung zum Logarithmus der Plasma-TENORMIN-Konzentration. Die Wirkung einer intravenösen Einzeldosis von 10 mg auf Belastungstachykardie ist nach 12 Stunden weitgehend abgeklungen, wohingegen die Betablocker-Aktivität von oralen Einzeldosen von 50 mg und 100 mg noch über 24 Stunden nach der Verabreichung nachweisbar ist. Wie jedoch für alle Betablocker gezeigt wurde, scheint die blutdrucksenkende Wirkung nicht von den Plasmaspiegeln abhängig zu sein.
Bei gesunden Probanden wurde die Beta1-Selektivität von TENORMIN 100 mg durch seine verringerte Fähigkeit gezeigt, die Beta2-vermittelte gefäßerweiternde Wirkung von Isoproterenol im Vergleich zu äquivalenten Beta-blockierenden Dosen von Propranolol umzukehren. Bei Asthmapatienten führte eine Dosis von TENORMIN 25 mg, die eine größere Wirkung auf die Herzfrequenz in Ruhe hatte als Propranolol, zu einem viel geringeren Anstieg des Atemwegswiderstands. In einem Placebo-kontrollierten Vergleich von etwa gleich wirksamen oralen Dosen mehrerer Betablocker führte TENORMIN zu einer signifikant geringeren Senkung des FEV1 als nichtselektive Betablocker wie Propranolol und hemmte im Gegensatz zu diesen Wirkstoffen die Bronchodilatation als Reaktion auf Isoproterenol nicht.
In Übereinstimmung mit seiner negativen chronotropen Wirkung aufgrund der Beta-Blockade des SA-Knotens erhöht TENORMIN 25 mg die Länge des Sinuszyklus und die Erholungszeit des Sinusknotens. Die Überleitung im AV-Knoten wird ebenfalls verlängert. TENORMIN 25 mg hat keine membranstabilisierende Wirkung, und eine Erhöhung der Dosis weit über die Betablockade hinaus senkt die myokardiale Kontraktilität nicht weiter. Mehrere Studien haben eine moderate (ca. 10 %) Zunahme des Schlagvolumens in Ruhe und unter Belastung gezeigt.
In kontrollierten klinischen Studien war TENORMIN, verabreicht als orale Einzeldosis, ein wirksames Antihypertonikum, das den Blutdruck über 24 Stunden senkte. TENORMIN 50 mg wurde in Kombination mit Diuretika vom Thiazidtyp untersucht, und die blutdrucksenkenden Wirkungen der Kombination sind ungefähr additiv. TENORMIN 50 mg ist auch kompatibel mit Methyldopa, Hydralazin und Prazosin, wobei jede Kombination zu einer stärkeren Blutdrucksenkung führt als die Einzelwirkstoffe. Der Dosisbereich von TENORMIN ist eng und eine Erhöhung der Dosis über 100 mg einmal täglich ist nicht mit einer verstärkten blutdrucksenkenden Wirkung verbunden. Die Mechanismen der blutdrucksenkenden Wirkung von Betablockern sind nicht bekannt. Mehrere mögliche Mechanismen wurden vorgeschlagen und umfassen: (1) kompetitiver Antagonismus von Katecholaminen an peripheren (insbesondere kardialen) adrenergen Neuronenstellen, was zu einer verringerten Herzleistung führt, (2) ein zentraler Effekt, der zu einem verringerten sympathischen Abfluss in die Peripherie führt, und (3 ) Unterdrückung der Reninaktivität. Die Ergebnisse aus Langzeitstudien haben keine Verringerung der blutdrucksenkenden Wirkung von TENORMIN 25 mg bei längerer Anwendung gezeigt.
Durch die Blockierung der positiven chronotropen und inotropen Wirkungen von Katecholaminen und durch die Senkung des Blutdrucks reduziert Atenolol im Allgemeinen den Sauerstoffbedarf des Herzens bei jeder gegebenen Anstrengung, was es für viele Patienten bei der Langzeitbehandlung von Angina pectoris nützlich macht. Andererseits kann Atenolol den Sauerstoffbedarf erhöhen, indem es die Länge der linksventrikulären Fasern und den enddiastolischen Druck erhöht, insbesondere bei Patienten mit Herzinsuffizienz.
In einer multizentrischen klinischen Studie (ISIS-1), die an 16.027 Patienten mit Verdacht auf Myokardinfarkt durchgeführt wurde, wurden Patienten, die sich innerhalb von 12 Stunden (Mittelwert = 5 Stunden) nach dem Einsetzen der Schmerzen vorstellten, entweder einer konventionellen Therapie plus TENORMIN (n = 8.037) oder randomisiert konventionelle Therapie allein (n = 7.990). Patienten mit einer Herzfrequenz von
Während des Behandlungszeitraums (Tage 0-7) betrug die vaskuläre Sterblichkeitsrate 3,89 % in der TENORMIN-Gruppe (313 Todesfälle) und 4,57 % in der Kontrollgruppe (365 Todesfälle). Dieser absolute Unterschied in den Raten, 0,68 %, ist statistisch signifikant auf dem Niveau P
Trotz des großen Umfangs der ISIS-1-Studie ist es nicht möglich, eindeutig Untergruppen von Patienten zu identifizieren, die am wahrscheinlichsten oder am wenigsten wahrscheinlich von einer frühen Behandlung mit Atenolol profitieren würden. Ein gutes klinisches Urteil legt jedoch nahe, dass Patienten, die zur Aufrechterhaltung eines angemessenen Herzzeitvolumens und Blutdrucks auf eine sympathische Stimulation angewiesen sind, keine guten Kandidaten für eine Betablockade sind. Tatsächlich spiegelte das Studienprotokoll diese Einschätzung wider, indem Patienten mit einem systolischen Blutdruck von dauerhaft unter 100 mm Hg ausgeschlossen wurden. Die Gesamtergebnisse der Studie sind mit der Möglichkeit vereinbar, dass Patienten mit einem grenzwertigen Blutdruck (weniger als 120 mm Hg systolisch), insbesondere wenn sie über 60 Jahre alt sind, weniger wahrscheinlich davon profitieren.
Der Mechanismus, durch den Atenolol das Überleben bei Patienten mit gesichertem oder vermutetem akutem Myokardinfarkt verbessert, ist unbekannt, wie dies bei anderen Betablockern in der Postinfarktsituation der Fall ist. Atenolol hat zusätzlich zu seinen Auswirkungen auf das Überleben andere klinische Vorteile gezeigt, einschließlich einer verringerten Häufigkeit ventrikulärer Extrasystolen, verringerter Brustschmerzen und verringerter Enzymerhöhung.
Atenolol Geriatrische Pharmakologie
Im Allgemeinen weisen ältere Patienten höhere Atenolol-Plasmaspiegel mit etwa 50 % niedrigeren Gesamt-Clearance-Werten als jüngere Patienten auf. Die Halbwertszeit ist bei älteren Menschen deutlich länger als bei jüngeren Probanden. Die Abnahme der Atenolol-Clearance folgt dem allgemeinen Trend, dass die Elimination renal ausgeschiedener Arzneimittel mit zunehmendem Alter abnimmt.
INFORMATIONEN ZUM PATIENTEN
Keine Informationen bereitgestellt. Bitte wende dich an die WARNUNGEN und VORSICHTSMASSNAHMEN Abschnitte.
Was ist Theo-24 200 mg und wie wird es angewendet?
Theo-24 200 mg ist ein verschreibungspflichtiges Medikament zur Behandlung der Symptome von Asthma, Bronchitis und Emphysem. Theo-24 200 mg kann allein oder mit anderen Medikamenten verwendet werden.
Theo-24 gehört zu einer Klasse von Medikamenten namens Xanthin-Derivate; Phosphodiesterase-Enzym-Inhibitoren, nicht selektiv.
Welche Nebenwirkungen kann Theo-24 200mg haben?
Theo-24 kann schwerwiegende Nebenwirkungen verursachen, einschließlich:
Suchen Sie sofort medizinische Hilfe auf, wenn Sie eines der oben aufgeführten Symptome haben.
Die häufigsten Nebenwirkungen von Theo-24 sind:
Teilen Sie dem Arzt mit, wenn Sie eine Nebenwirkung haben, die Sie stört oder die nicht abklingt.
Dies sind nicht alle möglichen Nebenwirkungen von Theo-24. Für weitere Informationen fragen Sie Ihren Arzt oder Apotheker.
Rufen Sie Ihren Arzt für medizinischen Rat zu Nebenwirkungen an. Sie können Nebenwirkungen der FDA unter 1-800-FDA-1088 melden.
BEZEICHNUNG
Theophyllin
Theophyllin wird strukturell als Methylxanthin klassifiziert. Es kommt als weißes, geruchloses, kristallines Pulver mit bitterem Geschmack vor. Wasserfreies Theophyllin hat den chemischen Namen 1H-Purin-2,6-dion, 3,7-dihydro-1,3-dimethyl- und wird durch die folgende Strukturformel dargestellt:
Die Summenformel von wasserfreiem Theophyllin ist C7H8N4O2 mit einem Molekulargewicht von 180,17.
Theo-24 (wasserfreie Theophyllin-Kapsel) ® ist als Kapseln erhältlich, die zur oralen Verabreichung bestimmt sind und 100 mg, 200 mg, 300 mg oder 400 mg wasserfreies Theophyllin pro Kapsel enthalten, in einer Formulierung mit verlängerter Freisetzung, die eine Dosierung über 24 Stunden ermöglicht Intervall für geeignete Patienten. Inaktive Inhaltsstoffe sind essbare Tinte (die synthetisches schwarzes Eisenoxid, FD&C Blau Nr. 1, FD&C Blau Nr. 2, FD&C Gelb Nr. 6, D&C Gelb Nr. 10, FD&C Rot Nr. 40 enthält), Ethylcellulose, Gelatine, pharmazeutische Glasur , kolloidales Siliciumdioxid, Stärke, Saccharose, Talk, Titandioxid und Farbstoffe: 100 mg – enthält FD&C Yellow Nr. 6; 200 mg-FD&C Rot Nr. 3 und D&C Gelb Nr. 10; 300 mg-FD&C Blau Nr. 1 und FD&C Rot Nr. 40; 400 mg-FD&C Red Nr. 40 und D&C Red Nr. 28.
Theo-24 (Wasserfreie Theophyllin-Kapsel) ® Kapseln mit verlängerter Freisetzung erfüllen den Drug Release Test 6, wie in der aktuellen USP-Monographie für Theophyllin-Kapseln mit verlängerter Freisetzung veröffentlicht.
INDIKATIONEN
Theophyllin ist indiziert zur Behandlung der Symptome und der reversiblen Atemwegsobstruktion im Zusammenhang mit chronischem Asthma und anderen chronischen Lungenerkrankungen, z. B. Emphysem und chronischer Bronchitis.
DOSIERUNG UND ANWENDUNG
allgemeine Überlegungen
Theo-24 (wasserfreie Theophyllin-Kapsel) ® ist, wie andere Theophyllin-Produkte mit verlängerter Freisetzung, für Patienten mit relativ anhaltenden oder wiederkehrenden Symptomen bestimmt, die einen therapeutischen Theophyllin-Serumspiegel aufrechterhalten müssen. Es ist nicht für Patienten bestimmt, die an einer akuten Episode von Bronchospasmus leiden (in Verbindung mit Asthma, chronischer Bronchitis oder Emphysem). Solche Patienten benötigen eine schnelle Linderung der Symptome und sollten mit einem Theophyllin-Präparat mit sofortiger Freisetzung oder intravenöser Verabreichung (oder anderen Bronchodilatatoren) und nicht mit Produkten mit verlängerter Freisetzung behandelt werden.
Patienten, die Theophyllin mit normaler oder langsamer Rate metabolisieren, sind vernünftige Kandidaten für die einmal tägliche Gabe von Theo-24 (wasserfreie Theophyllin-Kapsel) ® . Patienten, die Theophyllin schnell metabolisieren (z. B. junge Menschen, Raucher und einige nicht rauchende Erwachsene) und bei denen am Ende eines Dosierungsintervalls wiederholt Symptome auftreten, benötigen entweder einmal täglich erhöhte Dosen oder vorzugsweise eine bessere Kontrolle durch ein Zeitplan mit zweimal täglicher Dosierung. Bei Patienten, die erhöhte Tagesdosen benötigen, ist es wahrscheinlicher, dass sie relativ große Peak-Trough-Unterschiede erfahren und möglicherweise Kandidaten für eine zweimal tägliche Dosierung mit Theo-24 (wasserfreie Theophyllin-Kapsel) ® sind.
Die Patienten sollten angewiesen werden, dieses Medikament jeden Morgen ungefähr zur gleichen Zeit einzunehmen und die verschriebene Dosis nicht zu überschreiten.
Jüngste Studien deuten darauf hin, dass die nächtliche Einnahme von Theophyllin-Produkten mit verlängerter Wirkstofffreisetzung (nach dem Abendessen) zu Theophyllin-Serumkonzentrationen führt, die nicht mit denen identisch sind, die während der Wachstunden gemessen wurden, und durch frühe Talspiegel und verzögerte Höchstspiegel gekennzeichnet sein können. Dies scheint unabhängig davon aufzutreten, ob das Medikament als Produkt mit sofortiger Freisetzung, verlängerter Freisetzung oder intravenös verabreicht wird. Um dieses Phänomen zu vermeiden, wenn zwei Dosen pro Tag verschrieben werden, wird empfohlen, die zweite Dosis 10 bis 12 Stunden nach der morgendlichen Dosis und vor dem Abendessen zu geben.
Ernährung und Körperhaltung können zusammen mit Veränderungen im Zusammenhang mit dem zirkadianen Rhythmus die Resorptionsrate und/oder Clearance-Raten von Theophyllin aus Retard-Dosierungsformen beeinflussen, die nachts verabreicht werden. Die genaue Beziehung dieser und anderer Faktoren zu nächtlichen Serumkonzentrationen und die klinische Bedeutung solcher Befunde erfordern zusätzliche Studien. Daher wird das nicht empfohlen
Theo-24 (wasserfreie Theophyllin-Kapsel) ® (bei Anwendung als einmal täglich einzunehmendes Produkt) wird nachts verabreicht.
Patienten, die eine relativ hohe Dosis Theophyllin benötigen (d. h. eine Dosis von mindestens 900 mg oder 13 mg/kg, je nachdem, welcher Wert geringer ist), sollten Theo-24 (wasserfreie Theophyllin-Kapsel) ® nicht weniger als 1 Stunde vor einem High einnehmen -fetthaltige Mahlzeit, da dies zu einem signifikanten Anstieg des Spitzenserumspiegels und des Ausmaßes der Resorption von Theophyllin im Vergleich zur Einnahme im nüchternen Zustand führen kann (vgl VORSICHTSMASSNAHMEN, Wechselwirkungen zwischen Arzneimitteln und Lebensmitteln ).
Die Theophyllin-Spitzenkonzentration im Steady-State im Serum ist eine Funktion der Dosis, des Dosierungsintervalls und der Resorptions- und Clearance-Rate von Theophyllin beim einzelnen Patienten. Aufgrund deutlicher individueller Unterschiede in der Rate der Theophyllin-Clearance variiert die Dosis, die erforderlich ist, um eine Theophyllin-Spitzenkonzentration im Serum im Bereich von 10–20 mcg/ml zu erreichen, bei ansonsten ähnlichen Patienten um das Vierfache, wenn keine Faktoren bekannt sind, die die Theophyllin-Clearance verändern (z. 400-1600 mg/Tag bei Erwachsenen Die Dosis von Theophyllin muss auf der Grundlage der Messungen der Spitzenkonzentration von Theophyllin im Serum individualisiert werden, um eine Dosis zu erreichen, die einen maximalen potenziellen Nutzen bei minimalem Risiko von Nebenwirkungen bietet.
Vorübergehende koffeinähnliche Nebenwirkungen und übermäßige Serumkonzentrationen in langsamen Metabolisierern können bei den meisten Patienten vermieden werden, indem mit einer ausreichend niedrigen Dosis begonnen und die Dosis langsam in kleinen Schritten erhöht wird, wenn dies klinisch angezeigt ist (siehe ). Dosiserhöhungen sollten nur vorgenommen werden, wenn die vorherige Dosis gut vertragen wird, und in Abständen von mindestens 3 Tagen, damit die Theophyllin-Serumkonzentrationen den neuen Steady State erreichen können. Die Dosisanpassung sollte sich an der Messung der Theophyllinkonzentration im Serum orientieren (siehe VORSICHTSMASSNAHMEN, Labortests und DOSIERUNG UND VERABREICHUNG, Tabelle VI ). Gesundheitsdienstleister sollten Patienten und Pflegepersonal anweisen, jede Dosierung, die Nebenwirkungen verursacht, abzusetzen, die Medikation auszusetzen, bis diese Symptome verschwunden sind, und die Therapie dann mit einer niedrigeren, zuvor verträglichen Dosierung fortzusetzen (siehe WARNUNGEN ).
Wenn die Symptome des Patienten gut unter Kontrolle sind, gibt es keine offensichtlichen Nebenwirkungen und keine intervenierenden Faktoren, die die Dosierungsanforderungen verändern könnten (siehe WARNUNGEN und VORSICHTSMASSNAHMEN ), sollten die Theophyllin-Serumkonzentrationen bei schnell wachsenden Kindern in Abständen von 6 Monaten und bei allen anderen in jährlichen Abständen überwacht werden. Bei akut erkrankten Patienten sollten die Serumkonzentrationen von Theophyllin in kurzen Abständen, z. B. alle 24 Stunden, überwacht werden.
Theophyllin verteilt sich schlecht im Körperfett, daher sollte die mg/kg-Dosis auf der Grundlage des idealen Körpergewichts berechnet werden. Tabelle V enthält das Titrationsschema für die Theophyllin-Dosierung, das für Patienten in verschiedenen Altersgruppen und klinischen Situationen empfohlen wird. Tabelle VI enthält Empfehlungen für die Anpassung der Theophyllin-Dosierung basierend auf den Serum-Theophyllin-Konzentrationen. Bei der Anwendung dieser allgemeinen Dosierungsempfehlungen auf einzelne Patienten müssen die einzigartigen klinischen Merkmale jedes Patienten berücksichtigt werden. Im Allgemeinen sollten diese Empfehlungen als Obergrenze für Dosisanpassungen dienen, um das Risiko potenziell schwerwiegender unerwünschter Ereignisse im Zusammenhang mit unerwarteten starken Anstiegen der Theophyllinkonzentration im Serum zu verringern.
B. Patienten mit Risikofaktoren für eine beeinträchtigte Clearance, ältere Menschen (> 60 Jahre) und solche, bei denen es nicht möglich ist, die Theophyllinkonzentrationen im Serum zu überwachen:
Bei Kindern im Alter von 12 bis 15 Jahren sollte die endgültige Theophyllin-Dosis 16 mg/kg/Tag bis zu einem Maximum von 400 mg/Tag nicht überschreiten, wenn Risikofaktoren für eine verringerte Theophyllin-Clearance vorliegen (siehe WARNUNGEN ) oder wenn es nicht möglich ist, die Theophyllinkonzentrationen im Serum zu überwachen.
Bei Jugendlichen ≥ 16 Jahren und Erwachsenen, einschließlich älterer Menschen, sollte die endgültige Theophyllin-Dosis 400 mg/Tag nicht überschreiten, wenn Risikofaktoren für eine verringerte Theophyllin-Clearance vorliegen (siehe Abschnitt 4.4). WARNUNGEN ) oder wenn es nicht möglich ist, die Theophyllinkonzentrationen im Serum zu überwachen.
* Patienten mit schnellerem Metabolismus, die klinisch durch einen überdurchschnittlichen Dosisbedarf identifiziert wurden, sollten häufiger eine kleinere Dosis erhalten, um Durchbruchsymptome zu vermeiden, die aus niedrigen Talkonzentrationen vor der nächsten Dosis resultieren. Eine zuverlässig absorbierte Formulierung mit langsamer Freisetzung verringert Schwankungen und ermöglicht längere Dosierungsintervalle.
WIE GELIEFERT
Theo-24 (wasserfreie Theophyllin-Kapsel) ® (wasserfreies Theophyllin) wird in Kapseln mit verlängerter Freisetzung geliefert, die 100, 200, 300 oder 400 mg wasserfreies Theophyllin enthalten.
Theo-24 (wasserfreie Theophyllin-Kapsel) ® 100-mg-Kapseln sind gelb-orange und durchsichtig, mit den Markierungen Theo-24 (wasserfreie Theophyllin-Kapsel), 100 mg, ucb und 2832, geliefert als:
Lagerung
Unter 77 °F (25 °C) lagern.
FÜR MEDIZINISCHE INFORMATIONEN Kontakt: Abteilung für medizinische Angelegenheiten Telefon: (800) 477-7877, Fax: (770) 970-8859. Hergestellt für: UCB Pharma, Inc. Smyrna, GA 30080. von Pfizer Pharmaceuticals LLC Caguas, PR 00725. 04/2005.
NEBENWIRKUNGEN
Nebenwirkungen im Zusammenhang mit Theophyllin sind im Allgemeinen mild, wenn die Theophyllin-Spitzenkonzentration im Serum ÜBERDOSIERUNG ). Die vorübergehenden koffeinähnlichen Nebenwirkungen treten bei etwa 50 % der Patienten auf, wenn die Therapie mit Theophyllin mit höheren Dosen als den empfohlenen Anfangsdosen begonnen wird (z. B. > 300 mg/Tag bei Erwachsenen und > 12 mg/kg/Tag bei Kindern über 1 Jahr). das Alter). Zu Beginn einer Therapie mit Theophyllin können koffeinähnliche Nebenwirkungen das Patientenverhalten vorübergehend verändern, insbesondere bei Kindern im Schulalter, aber diese Reaktion hält selten an. Der Beginn einer Therapie mit Theophyllin in niedriger Dosis mit anschließender langsamer Titration bis zu einer vorbestimmten altersabhängigen Höchstdosis wird die Häufigkeit dieser vorübergehenden Nebenwirkungen signifikant verringern (siehe DOSIERUNG UND VERABREICHUNG, Tabelle V ). Bei einem kleinen Prozentsatz der Patienten (
Andere Nebenwirkungen, die bei Theophyllin-Serumkonzentrationen
WECHSELWIRKUNGEN MIT ANDEREN MEDIKAMENTEN
Wechselwirkungen zwischen Arzneimitteln und Arzneimitteln
Theophyllin interagiert mit einer Vielzahl von Medikamenten. Die Wechselwirkung kann pharmakodynamischer Natur sein, dh Veränderungen in der therapeutischen Reaktion auf Theophyllin oder ein anderes Arzneimittel oder das Auftreten von Nebenwirkungen ohne Veränderung der Theophyllinkonzentration im Serum. Häufiger ist jedoch die Wechselwirkung pharmakokinetischer Natur, dh die Geschwindigkeit der Theophyllin-Clearance wird durch ein anderes Medikament verändert, was zu erhöhten oder verringerten Serum-Theophyllin-Konzentrationen führt. Theophyllin verändert die Pharmakokinetik anderer Arzneimittel nur selten.
Die in Tabelle II aufgeführten Arzneimittel haben das Potenzial, klinisch signifikante pharmakodynamische oder pharmakokinetische Wechselwirkungen mit Theophyllin hervorzurufen. Die Informationen in der Spalte „Wirkung“ von Tabelle II gehen davon aus, dass das interagierende Arzneimittel zu einer stationären Therapie mit Theophyllin hinzugefügt wird. Wenn eine Behandlung mit Theophyllin bei einem Patienten begonnen wird, der bereits ein Arzneimittel einnimmt, das die Theophyllin-Clearance hemmt (z. B. Cimetidin, Erythromycin), ist die zum Erreichen einer therapeutischen Theophyllin-Serumkonzentration erforderliche Theophyllin-Dosis geringer. Umgekehrt, wenn eine Behandlung mit Theophyllin bei einem Patienten begonnen wird, der bereits ein Medikament einnimmt, das die Theophyllin-Clearance erhöht (z. B. Rifampin), ist die Theophyllin-Dosis, die erforderlich ist, um eine therapeutische Theophyllin-Serumkonzentration zu erreichen, höher. Das Absetzen eines gleichzeitig verabreichten Arzneimittels, das die Theophyllin-Clearance erhöht, führt zu einer Akkumulation von Theophyllin auf potenziell toxische Niveaus, es sei denn, die Theophyllin-Dosis wird angemessen reduziert. Das Absetzen eines gleichzeitig verabreichten Arzneimittels, das die Theophyllin-Clearance hemmt, führt zu verringerten Serum-Theophyllin-Konzentrationen, es sei denn, die Theophyllin-Dosis wird angemessen erhöht.
Für die in Tabelle III aufgeführten Arzneimittel wurde entweder dokumentiert, dass sie nicht mit Theophyllin interagieren, oder sie erzeugen keine klinisch signifikante Wechselwirkung (dh
Die Liste der Medikamente in Tabelle II ist auf dem Stand von Juni 2004. Die Liste der Medikamente in Tabelle III ist auf dem Stand vom 2. Januar 1996. Für Theophyllin werden ständig neue Wechselwirkungen gemeldet, insbesondere mit neuen chemischen Substanzen. Das medizinische Fachpersonal sollte nicht davon ausgehen, dass ein Medikament nicht mit Theophyllin interagiert, wenn es nicht in Tabelle II aufgeführt ist. Vor der Zugabe eines neu erhältlichen Arzneimittels zu einem Patienten, der Theophyllin erhält, sollte die Packungsbeilage des neuen Arzneimittels und/oder die medizinische Literatur konsultiert werden, um festzustellen, ob eine Wechselwirkung zwischen dem neuen Arzneimittel und Theophyllin berichtet wurde.
Wechselwirkungen zwischen Arzneimitteln und Nahrungsmitteln
Einnahme von Theo-24 (wasserfreie Theophyllin-Kapsel) ® weniger als eine Stunde vor einer fettreichen Mahlzeit, wie z. B. 8 Unzen Vollmilch, 2 Spiegeleier, 2 Speckstreifen, 2 Unzen Kartoffelpüree und 2 Scheiben Buttertoast (ca. 985 Kalorien, einschließlich ca. 71 g Fett) kann zu einem signifikanten Anstieg des Spitzenserumspiegels und des Ausmaßes der Resorption von Theophyllin im Vergleich zur Verabreichung im nüchternen Zustand führen. In einigen Fällen (insbesondere bei Dosen von 900 mg oder mehr, die weniger als eine Stunde vor einer fettreichen Mahlzeit eingenommen werden) können die Serumspiegel von Theophyllin den Wert von 20 mcg/ml überschreiten, oberhalb dessen eine Theophyllin-Toxizität wahrscheinlicher ist.
Die Wirkung anderer Medikamente auf Theophyllin-Serumkonzentrationsmessungen
Die meisten klinisch verwendeten Serum-Theophyllin-Assays sind Immunoassays, die für Theophyllin spezifisch sind. Andere Xanthine wie Koffein, Dyphyllin und Pentoxifyllin werden von diesen Assays nicht nachgewiesen. Einige Medikamente (z. B. Cefazolin, Cephalothin) können jedoch bestimmte HPLC-Techniken stören. Koffein- und Xanthin-Metabolite bei Neugeborenen oder Patienten mit Nierenfunktionsstörungen können dazu führen, dass die Messwerte einiger Trockenreagenz-Büromethoden höher sind als die tatsächliche Serum-Theophyllin-Konzentration.
WARNUNGEN
Gleichzeitige Erkrankung
Theophyllin sollte bei Patienten mit den folgenden klinischen Zuständen aufgrund des erhöhten Risikos einer Exazerbation der Begleiterkrankung mit äußerster Vorsicht angewendet werden:
Aktive Magengeschwüre Krampfanfälle Herzrhythmusstörungen (ohne Bradyarrhythmien)
Bedingungen, die die Theophyllin-Clearance reduzieren
Es gibt mehrere leicht identifizierbare Ursachen für eine verringerte Theophyllin-Clearance. Wenn die tägliche Gesamtdosis bei Vorliegen dieser Risikofaktoren nicht angemessen reduziert wird, kann eine schwere und potenziell tödliche Theophyllin-Toxizität auftreten. Nutzen und Risiken der Anwendung von Theophyllin und die Notwendigkeit einer intensiveren Überwachung der Theophyllin-Serumkonzentrationen bei Patienten mit den folgenden Risikofaktoren müssen sorgfältig abgewogen werden:
Das Alter
Neugeborene (Termine und Frühgeborene) Kinder 60 Jahre)
Begleiterkrankungen
akutes Lungenödem dekompensierte Herzinsuffizienz Cor-pulmonale Fieber; ≥ 102 °F für 24 Stunden oder länger; oder geringere Temperaturerhöhungen über längere Zeit Hypothyreose Lebererkrankung; Zirrhose, akute Hepatitis Eingeschränkte Nierenfunktion bei Säuglingen
Raucherentwöhnung
Wechselwirkungen mit anderen Medikamenten
Hinzufügen eines Arzneimittels, das den Metabolismus von Theophyllin hemmt (z. B. Cimetidin, Erythromycin, Tacrin) oder Absetzen eines gleichzeitig verabreichten Arzneimittels, das den Metabolismus von Theophyllin verstärkt (z. B. Carbamazepin, Rifampin) (siehe VORSICHTSMASSNAHMEN: WECHSELWIRKUNGEN MIT ARZNEIMITTELN, Tabelle II ).
Wenn Anzeichen oder Symptome einer Theophyllin-Toxizität vorhanden sind
Wenn ein Patient, der Theophyllin erhält, Übelkeit oder Erbrechen entwickelt, insbesondere wiederholtes Erbrechen, oder andere Anzeichen oder Symptome, die mit einer Theophyllin-Toxizität vereinbar sind (auch wenn eine andere Ursache vermutet wird), sollten zusätzliche Theophyllin-Dosen ausgesetzt und sofort die Theophyllin-Serumkonzentration gemessen werden. Die Patienten sollten angewiesen werden, keine Dosierung fortzusetzen, die Nebenwirkungen verursacht, und nachfolgende Dosen auszusetzen, bis die Symptome abgeklungen sind; zu diesem Zeitpunkt kann das medizinische Fachpersonal den Patienten anweisen, das Medikament mit einer niedrigeren Dosierung wieder aufzunehmen (siehe Abschnitt 4.4). DOSIERUNG UND VERABREICHUNG, Dosierungsrichtlinien, Tabelle VI ).
Dosierung erhöht
Erhöhungen der Theophyllin-Dosis sollten nicht als Reaktion auf eine akute Verschlimmerung der Symptome einer chronischen Lungenerkrankung erfolgen, da Theophyllin unter diesen Umständen einen geringen Zusatznutzen gegenüber inhalativen Beta2-selektiven Agonisten und systemisch verabreichten Kortikosteroiden bietet und das Risiko von Nebenwirkungen erhöht. Eine Theophyllin-Spitzenspiegel im Steady State sollte gemessen werden, bevor die Dosis als Reaktion auf anhaltende chronische Symptome erhöht wird, um festzustellen, ob eine Dosiserhöhung sicher ist. Bevor die Theophyllin-Dosis aufgrund einer niedrigen Serumkonzentration erhöht wird, sollte das medizinische Fachpersonal prüfen, ob die Blutprobe im Verhältnis zur Dosis zu einem angemessenen Zeitpunkt entnommen wurde und ob sich der Patient an das verschriebene Regime gehalten hat (siehe Abschnitt 4.4). VORSICHTSMASSNAHMEN, Labortests ).
Da die Geschwindigkeit der Theophyllin-Clearance dosisabhängig sein kann (dh Steady-State-Serumkonzentrationen können überproportional zur Dosiserhöhung ansteigen), sollte eine Dosiserhöhung auf der Grundlage einer subtherapeutischen Messung der Serumkonzentration konservativ erfolgen. Im Allgemeinen verringert eine Begrenzung der Dosiserhöhung auf etwa 25 % der vorherigen Gesamttagesdosis das Risiko eines unbeabsichtigten übermäßigen Anstiegs der Theophyllinkonzentration im Serum (siehe DOSIERUNG UND VERABREICHUNG, Tabelle VI ).
VORSICHTSMASSNAHMEN
Allgemein
Vor Beginn der Theophyllin-Therapie, vor Erhöhungen der Theophyllin-Dosis und während der Nachsorge sollten die verschiedenen Wechselwirkungen mit Arzneimitteln und physiologischen Zuständen, die die Theophyllin-Clearance verändern können und eine Dosisanpassung erfordern, sorgfältig abgewogen werden (siehe WARNUNGEN ). Die zu Beginn der Therapie gewählte Dosis von Theophyllin sollte niedrig sein und wenn geduldet, langsam über einen Zeitraum von einer Woche oder länger erhöht, wobei die endgültige Dosis von der Überwachung der Theophyllinkonzentrationen im Serum und dem klinischen Ansprechen des Patienten bestimmt wird (siehe DOSIERUNG UND VERABREICHUNG, Tabelle V ).
Überwachung der Theophyllinkonzentrationen im Serum
Serum-Theophyllin-Konzentrationsmessungen sind leicht verfügbar und sollten verwendet werden, um zu bestimmen, ob die Dosierung angemessen ist. Insbesondere sollte die Theophyllinkonzentration im Serum wie folgt gemessen werden:
Um eine Dosiserhöhung anzuleiten, sollte die Blutprobe zum Zeitpunkt der erwarteten maximalen Theophyllin-Serumkonzentration entnommen werden; 12 Stunden nach einer Dosis im Steady State (erwarteter Theophyllin-Spitzenkonzentrationsbereich im Serum liegt zwischen 5 – 15 µg/ml). Bei den meisten Patienten wird der Steady-State nach 3-tägiger Einnahme erreicht, wenn keine Dosis ausgelassen, keine zusätzliche Dosis hinzugefügt und keine Dosis in ungleichen Abständen eingenommen wurde. Eine Talkonzentration (dh am Ende des Dosierungsintervalls) liefert keine zusätzlichen nützlichen Informationen und kann zu einer unangemessenen Dosiserhöhung führen, da die Spitzenkonzentration von Theophyllin im Serum zwei- oder mehrfach höher sein kann als die Talkonzentration bei einer Formulierung mit verlängerter Freisetzung . Wenn die Serumprobe mehr oder weniger als zwölf (12) Stunden nach der Dosis entnommen wird, müssen die Ergebnisse mit Vorsicht interpretiert werden, da die Konzentration möglicherweise nicht die Spitzenkonzentration widerspiegelt. Wenn dagegen Anzeichen oder Symptome einer Theophyllin-Toxizität vorliegen, sollte die Serumprobe so schnell wie möglich entnommen, sofort analysiert und das Ergebnis unverzüglich dem medizinischen Fachpersonal mitgeteilt werden. Bei Patienten, bei denen eine verminderte Serumproteinbindung vermutet wird (z. B. Zirrhose, Frauen im dritten Trimenon der Schwangerschaft), sollte die Konzentration von ungebundenem Theophyllin gemessen und die Dosis angepasst werden, um eine ungebundene Konzentration von 6-12 µg/ml Speichelkonzentration zu erreichen von Theophyllin kann ohne spezielle Techniken nicht zuverlässig verwendet werden, um die Dosierung anzupassen.
Auswirkungen auf Labortests
Aufgrund seiner pharmakologischen Wirkung erhöht Theophyllin bei Serumkonzentrationen im Bereich von 10–20 mcg/ml leicht die Plasmaglukose (von einem Mittelwert von 88 mg % auf 98 mg %), die Harnsäure (von einem Mittelwert von 4 mg/dl bis 6 mg/dL), freie Fettsäuren (von einem Mittelwert von 451 µEq/L bis 800 μEq/L), Gesamtcholesterin (von einem Mittelwert von 140 vs. 160 mg/dL), HDL (von einem Mittelwert von 36 bis 50 mg /dL), HDL/LDL-Verhältnis (von einem Mittelwert von 0,5 bis 0,7) und Ausscheidung von freiem Cortisol im Urin (von einem Mittelwert von 44 bis 63 mcg/24 h) Theophyllin bei Serumkonzentrationen im Bereich von 10–20 mcg/ml kann auch die Serumkonzentrationen von Triiodthyronin vorübergehend verringern (144 vor, 131 nach einer Woche und 142 ng/dl nach 4 Wochen Theophyllin).Die klinische Bedeutung dieser Veränderungen sollte gegen den potenziellen therapeutischen Nutzen von Theophyllin bei einzelnen Patienten abgewogen werden.
Karzinogenese, Mutagenese und Beeinträchtigung der Fruchtbarkeit
Langzeitstudien zur Karzinogenität wurden an Mäusen (orale Dosen 30-150 mg/kg) und Ratten (orale Dosen 5-75 mg/kg) durchgeführt. Ergebnisse stehen noch aus.
Theophyllin wurde in Ames-Salmonellen-, In-vivo- und In-vitro-Zytogenetik-, Mikronukleus- und Eierstock-Testsystemen des chinesischen Hamsters untersucht und hat sich nicht als genotoxisch erwiesen.
In einer 14-wöchigen kontinuierlichen Zuchtstudie beeinträchtigte Theophyllin, das Paarungspaaren von B6C3F1-Mäusen in oralen Dosen von 120, 270 und 500 mg/kg (ca B. durch eine Abnahme der Anzahl lebender Jungtiere pro Wurf, eine Abnahme der mittleren Anzahl von Würfen pro fruchtbarem Paar und eine Verlängerung der Tragzeit bei der hohen Dosis sowie eine Abnahme des Anteils lebend geborener Jungtiere bei der mittleren und hohen Dosis. In 13-wöchigen Toxizitätsstudien wurde Theophyllin F344-Ratten und B6C3F1-Mäusen in oralen Dosen von 40-300 mg/kg (etwa das 2,0-fache der Humandosis auf mg/m2-Basis) verabreicht. Bei der hohen Dosis wurde bei beiden Spezies eine systemische Toxizität beobachtet, einschließlich einer Abnahme des Hodengewichts.
Schwangerschaft
Kategorie C
In Studien, in denen trächtige Mäuse, Ratten und Kaninchen während der Organogenese behandelt wurden, hatte Theophyllin teratogene Wirkungen.
In Studien mit Mäusen führte eine intraperitoneale Einzeldosis von 100 mg/kg und mehr (ungefähr gleich der maximal empfohlenen oralen Dosis für Erwachsene auf mg/m2-Basis) während der Organogenese zu Gaumenspalten und Fingerabnormalitäten. Mikromelie, Mikrognathie, Klumpfuß, subkutanes Hämatom, offene Augenlider und Embryoletalität wurden bei Dosen beobachtet, die ungefähr dem Zweifachen der maximal empfohlenen oralen Dosis für Erwachsene auf mg/m2-Basis entsprachen.
In einer Studie mit Ratten, denen von der Empfängnis bis zur Organogenese eine Dosis verabreicht wurde, führte eine orale Dosis von 150 mg/kg/Tag (etwa das Zweifache der maximal empfohlenen oralen Dosis für Erwachsene auf mg/m2-Basis) zu digitalen Anomalien. Embryoletalität wurde bei einer subkutanen Dosis von 200 mg/kg/Tag beobachtet (etwa das 4-fache der maximal empfohlenen oralen Dosis für Erwachsene auf mg/m2-Basis).
In einer Studie, in der trächtige Kaninchen während der gesamten Organogenese verabreicht wurden, führte eine intravenöse Dosis von 60 mg/kg/Tag (ungefähr das Zweifache der maximal empfohlenen oralen Dosis für Erwachsene auf mg/m2-Basis) zum Tod eines Rehs und klinisch Anzeichen bei anderen, verursachte eine Gaumenspalte und war embryoletal. Dosen ab 15 mg/kg/Tag (weniger als die maximal empfohlene orale Dosis für Erwachsene auf mg/m2-Basis) erhöhten die Inzidenz von Skelettvariationen.
Es liegen keine adäquaten und gut kontrollierten Studien bei Schwangeren vor. Theophyllin sollte während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen das potenzielle Risiko für den Fötus rechtfertigt.
Stillende Mutter
Theophyllin wird in die Muttermilch ausgeschieden und kann bei gestillten Säuglingen Reizbarkeit oder andere Anzeichen einer leichten Toxizität hervorrufen. Die Konzentration von Theophyllin in der Muttermilch entspricht etwa der mütterlichen Serumkonzentration. Ein Säugling, der einen Liter Muttermilch mit 10–20 mcg/ml Theophyllin pro Tag zu sich nimmt, erhält wahrscheinlich 10–20 mg Theophyllin pro Tag. Schwerwiegende Nebenwirkungen beim Säugling sind unwahrscheinlich, es sei denn, die Mutter hat toxische Serumkonzentrationen von Theophyllin.
Pädiatrische Verwendung
Theophyllin ist für die zugelassenen Indikationen bei pädiatrischen Patienten sicher und wirksam (siehe INDIKATIONEN ). Die Erhaltungsdosis von Theophyllin muss bei pädiatrischen Patienten mit Vorsicht ausgewählt werden, da die Geschwindigkeit der Theophyllin-Clearance über die Altersspanne von Neugeborenen bis hin zu Jugendlichen sehr unterschiedlich ist (siehe KLINISCHE PHARMAKOLOGIE, Tabelle I, WARNHINWEISE, und DOSIERUNG UND VERABREICHUNG, Tabelle V ). Aufgrund der Unreife der Theophyllin-Stoffwechselwege bei Säuglingen unter einem Jahr ist bei der Verschreibung von Theophyllin an pädiatrische Patienten dieser Altersgruppe eine besondere Aufmerksamkeit bei der Dosierungsauswahl und häufigen Überwachung der Theophyllin-Serumkonzentrationen erforderlich.
Geriatrische Verwendung
Ältere Patienten haben aufgrund altersbedingter pharmakokinetischer und pharmakodynamischer Veränderungen ein signifikant höheres Risiko, eine schwere Toxizität durch Theophyllin zu erfahren, als jüngere Patienten. Die Clearance von Theophyllin ist bei gesunden älteren Erwachsenen (> 60 Jahre) im Vergleich zu gesunden jungen Erwachsenen um durchschnittlich 30 % verringert. Die Clearance von Theophyllin kann durch bei älteren Menschen vorherrschende Begleiterkrankungen weiter verringert werden, die die Clearance dieses Arzneimittels weiter beeinträchtigen und das Potenzial haben, die Serumspiegel und die potenzielle Toxizität zu erhöhen. Zu diesen Erkrankungen gehören eingeschränkte Nierenfunktion, chronisch obstruktive Lungenerkrankung, dekompensierte Herzinsuffizienz, Lebererkrankung und eine erhöhte Prävalenz der Einnahme bestimmter Medikamente (siehe VORSICHTSMASSNAHMEN: WECHSELWIRKUNGEN MIT ARZNEIMITTELN mit dem Potenzial für pharmakokinetische und pharmakodynamische Wechselwirkungen. Die Proteinbindung kann bei älteren Menschen verringert sein, was zu einem erhöhten Anteil der gesamten Theophyllin-Serumkonzentration in der pharmakologisch aktiven ungebundenen Form führt. Ältere Patienten scheinen auch empfindlicher auf die toxischen Wirkungen von Theophyllin nach chronischer Überdosierung zu reagieren als jüngere Patienten. Bei älteren Patienten ist eine sorgfältige Dosisreduktion und eine häufige Überwachung der Theophyllinkonzentrationen im Serum erforderlich (siehe VORSICHTSMASSNAHMEN, Überwachung der Theophyllinkonzentrationen im Serum, und DOSIERUNG UND ANWENDUNG ). Die maximale Tagesdosis von Theophyllin bei Patienten über 60 Jahren sollte normalerweise 400 mg/Tag nicht überschreiten, es sei denn, der Patient ist weiterhin symptomatisch und die Theophyllin-Spitzenkonzentration im Steady-State im Serum beträgt DOSIERUNG UND ANWENDUNG ). Theophyllin-Dosen von mehr als 400 mg/Tag sollten bei älteren Patienten mit Vorsicht verschrieben werden.
ÜBERDOSIS
Allgemein
Die Chronizität und das Muster einer Theophyllin-Überdosierung beeinflussen signifikant die klinischen Manifestationen der Toxizität, das Management und das Ergebnis. Es gibt zwei häufige Erscheinungsformen: (1) akute Überdosierung, d. h. Einnahme einer einzelnen großen übermäßigen Dosis (> 10 mg/kg), wie sie im Zusammenhang mit einem Suizidversuch oder einem isolierten Medikationsfehler auftritt, und (2) chronische Überdosierung, d. h , Einnahme wiederholter Dosen, die für die Rate der Theophyllin-Clearance des Patienten zu hoch sind. Zu den häufigsten Ursachen einer chronischen Theophyllin-Überdosierung gehören Dosierungsfehler des Patienten oder des Pflegepersonals, die Verschreibung einer zu hohen Dosis oder einer normalen Dosis durch medizinisches Fachpersonal in Gegenwart von Faktoren, von denen bekannt ist, dass sie die Rate der Theophyllin-Clearance verringern, und eine Erhöhung der Dosis als Reaktion auf eine Verschlimmerung der Symptome ohne vorherige Messung der Theophyllinkonzentration im Serum, um festzustellen, ob eine Dosiserhöhung sicher ist.
Eine schwere Toxizität durch eine Theophyllin-Überdosierung ist ein relativ seltenes Ereignis. In einer Gesundheitsversorgungsorganisation betrug die Häufigkeit von Krankenhauseinweisungen wegen chronischer Überdosierung von Theophyllin etwa 1 pro 1000 Personenjahre Exposition. In einer anderen Studie lagen von 6000 Blutproben, die zur Messung der Theophyllinkonzentration im Serum aus irgendeinem Grund von in einer Notaufnahme behandelten Patienten entnommen wurden, 7 % im Bereich von 20–30 µg/ml und 3 % > 30 µg/ml. Etwa zwei Drittel der Patienten mit Theophyllin-Serumkonzentrationen im Bereich von 20-30 µg/ml zeigten eine oder mehrere Toxizitätserscheinungen, während > 90 % der Patienten mit Theophyllin-Serumkonzentrationen > 30 µg/ml klinisch vergiftet waren. In ähnlicher Weise wird in anderen Berichten eine ernsthafte Toxizität von Theophyllin hauptsächlich bei Serumkonzentrationen > 30 mcg/ml gesehen.
Mehrere Studien haben die klinischen Manifestationen einer Theophyllin-Überdosierung beschrieben und versucht, die Faktoren zu bestimmen, die eine lebensbedrohliche Toxizität vorhersagen. Im Allgemeinen ist die Wahrscheinlichkeit von Krampfanfällen bei Patienten mit akuter Überdosierung geringer als bei Patienten mit chronischer Überdosierung, es sei denn, die Theophyllin-Spitzenkonzentration im Serum beträgt > 100 µg/ml. Nach einer chronischen Überdosierung können bei Theophyllin-Serumkonzentrationen > 30 mcg/ml generalisierte Krampfanfälle, lebensbedrohliche Herzrhythmusstörungen und Tod auftreten. Der Schweregrad der Toxizität nach chronischer Überdosierung korreliert stärker mit dem Alter des Patienten als mit der Theophyllin-Spitzenkonzentration im Serum; Patienten > 60 Jahre haben das größte Risiko für schwere Toxizität und Mortalität nach einer chronischen Überdosierung. Vorbestehende oder gleichzeitig bestehende Erkrankungen können auch die Anfälligkeit eines Patienten für eine bestimmte toxische Manifestation signifikant erhöhen, z. B. haben Patienten mit neurologischen Erkrankungen ein erhöhtes Risiko für Krampfanfälle und Patienten mit Herzerkrankungen haben ein erhöhtes Risiko für Herzrhythmusstörungen bei einem gegebenen Serumtheophyllin Konzentration im Vergleich zu Patienten ohne Grunderkrankung.
Die Häufigkeit verschiedener berichteter Manifestationen einer Theophyllin-Überdosierung nach Art der Überdosierung sind in Tabelle IV aufgeführt.
Andere Manifestationen der Theophyllin-Toxizität umfassen Erhöhungen des Serumkalziums, der Kreatinkinase, der Myoglobin- und Leukozytenzahl, Abnahmen des Serumphosphats und -magnesiums, akuten Myokardinfarkt und Harnverhalt bei Männern mit obstruktiver Uropathie. Krampfanfälle im Zusammenhang mit Serum-Theophyllinkonzentrationen > 30 mcg/ml sind oft resistent gegen eine antikonvulsive Therapie und können zu irreversiblen Hirnschäden führen, wenn sie nicht schnell kontrolliert werden. Der Tod durch Theophyllin-Toxizität ist meistens Folge eines kardiorespiratorischen Stillstands und/oder einer hypoxischen Enzephalopathie nach anhaltenden generalisierten Krampfanfällen oder hartnäckigen Herzrhythmusstörungen, die eine hämodynamische Beeinträchtigung verursachen.
Überdosierungsmanagement
Allgemeine Empfehlungen für Patienten mit Symptomen einer Theophyllin-Überdosierung oder Theophyllin-Serumkonzentrationen > 30 mcg/ml (Hinweis: Die Theophyllin-Serumkonzentrationen können nach Vorstellung des Patienten in ärztlicher Behandlung weiter ansteigen.)